Chronic Bronchitis, Emphysema, and Airways Obstruction
Definition
Chronic obstructive pulmonary disease (COPD) is the name of a group of chronic and slowly progressive respiratory disorders characterized by reduced maximal expiratory flow during forced exhalation. Most of the airflow obstruction is fixed, but a variable degree of reversibility and bronchial hyperreactivity may be seen. COPD may coexist with asthma and, when abnormal airway reactivity is present, differentiation between these disorders can be challenging. COPD comprises emphysema and chronic bronchitis, two distinct processes, although most often present in combination. The definition excludes other causes of chronic airflow obstruction such as cystic fibrosis (Chap. 257), bronchiolitis obliterans (Chap. 259), and bronchiectasis (Chap. 256).
Emphysema is defined anatomically as a permanent and destructive enlargement of airspaces distal to the terminal bronchioles without obvious fibrosis and with loss of normal architecture. Chronic bronchitis is defined clinically as the presence of a cough productive of sputum not attributable to other causes on most days for at least 3 months over 2 consecutive years. Chronic bronchitis may be present in the absence of airflow limitation, but COPD always involves clinically significant airflow limitation.
Epidemiology
COPD is a common medical problem affecting an estimated 16 million Americans. Males are more frequently affected than females, and Caucasians more frequently than African Americans. There is a higher prevalence of COPD among persons with a lower socioeconomic status and in those with a history of low birth weight. COPD is the fourth leading cause of death in the United States and is the only one of the 10 leading causes of death for which mortality rates are still rising. Prevalence peaks in the seventh and eighth decades, then levels off, largely due to mortality.
Disease Mechanisms
Pathogenesis
COPD evolves from an inflammatory process involving the airways and distal airspaces. Increased activity of oxidants combined with decreased activity of antioxidants, termed oxidative stress, have been implicated in the development of inflammation and COPD. Cigarette smoke produces high concentrations of oxygen free radicals including superoxide, hydrogen peroxide, and hypochlorous acid. Cigarette smoke is an independent source of Fe2+, releases Fe2+ from ferritin, and catalyzes the formation of the highly active hydroxyl radical from O2- and H2O2 by eosinophils, neutrophils, and alveolar macrophages. Cigarette tar contains nitric oxide and induces nitric oxide synthase. In the presence of oxidants, NO is metabolized to cytotoxic peroxynitrates. In order for elastase to degrade elastin, 1 antitrypsin a 1AT) must be inactivated. Cigarette smoke, oxidants, activated neutrophils, and type II alveolar pneumocytes are all capable of inactivating a 1AT as well as matrix metalloproteinase inhibitors. Oxidant stress is also capable of inducing mucus hypersecretion. Cigarette smoke also acts as a chemoattractant and upregulates adhesion molecules. Smoke increases neutrophil transit time through the pulmonary circulation, increases adhesion, and decreases deformability. Smoke and elastase both increase the expression of the proinflammatory nuclear transcription factor B (NfB) as well as interleukin 8, a chemokine found to be elevated in COPD patients, that recruits neutrophils, basophils, eosinophils, and T lymphocytes.
The submucosa of the small airway in patients with COPD has increased numbers of CD8 lymphocytes and eosinophils, macrophages, and mast cells. Neutrophils are increased in smokers, but their numbers do not correlate with the presence of airflow obstruction. Patients with chronic airflow obstruction show higher levels of myeloperoxidase and eosinophilic cationic protein than do patients with normal airflow. Macrophages and mast cells produce transforming growth factor (TGF-), a peptide related to fibrogenesis. Patients with chronic airflow obstruction show a twofold elevation of TGF- in lavage liquid; the amount of TGF- shows a significant negative correlation with the forced expiratory volume in 1 s (FEV1). Smoke also leads to lipid peroxidation and to DNA damage. Widespread point mutations of the p53 gene locus have been identified in patients with lung cancer and precancerous dysplasia. These may predispose to the development of lung cancer.
Risk Factors
COPD is characterized by a reduced FEV1 and an accelerated rate of decline of FEV1. The reduction in FEV1 can occur by any of three pathways: (1) impaired childhood growth and development, with a lower peak in early adulthood and a normal rate of decline with aging (e.g., early childhood infection and passive smoke exposure); (2) normal growth and development with a premature peak but normal subsequent decline (e.g., asthma and passive smoking); and (3) normal growth and development and peak with accelerated decline (e.g., active smoking and, to a lesser degree, environmental exposures).
Smoking
Cigarette smoking is the most commonly identified correlate with both chronic bronchitis during life and extent of emphysema at postmortem. The prevalence of COPD shows a dose-response relationship with the number of pack-years of tobacco consumed. Some 90% of all COPD patients are current or former tobacco smokers. Experimental studies have shown that prolonged cigarette smoking impairs respiratory epithelial ciliary movement, inhibits function of alveolar macrophages, and leads to hypertrophy and hyperplasia of mucus-secreting glands; massive exposure in dogs can produce emphysematous changes. Cigarette smoke also inhibits antiproteases and causes polymorphonuclear leukocytes to release proteolytic enzymes acutely. Cigarette smoke can produce an acute increase in airways resistance due to vagally mediated smooth-muscle constriction by stimulating submucosal irritant receptors. Increased airways responsiveness is associated with more rapid progression in patients with chronic airways obstruction. Obstruction of small airways is the earliest demonstrable mechanical defect in young cigarette smokers and may disappear completely after cessation of smoking.
Although smoking cessation does not result in complete reversal of more pronounced obstruction, there is a significant slowing of the decline in lung function in all smokers who give up cigarettes. Passive exposure to tobacco smoke correlates with respiratory symptoms such as cough, wheeze, and sputum production. Not only is cigarette smoking the most common single factor leading to chronic airways obstruction, it also adds to the effects of every other contributory factor to be discussed below.
Air Pollution
The incidence and mortality rates of both chronic bronchitis and emphysema may be higher in heavily industrialized urban areas. Exacerbations of bronchitis are clearly related to periods of heavy pollution with sulfur dioxide (SO2) and particulate matter. While nitrogen dioxide (NO2) can produce small-airways obstruction (bronchiolitis) in experimental animals exposed to high concentrations, there are no data convincingly implicating NO2, at even the highest pollutant levels, in the pathogenesis or worsening of airways obstruction in humans.
Occupation
Chronic bronchitis is more prevalent in workers who engage in occupations exposing them to either inorganic or organic dusts or to noxious gases. Epidemiologic surveys have succeeded in demonstrating an accelerated decline in lung function in many such workers-e.g., workers in plastics plants exposed to toluene diisocyanate, and carding room workers in cotton mills (Chap. 254)-suggesting that their occupational exposure contributes to their future disability.
Infection
Morbidity, mortality, and frequency of acute respiratory illnesses are higher in patients with chronic bronchitis. Many attempts have been made to relate these illnesses to infection with viruses, mycoplasmas, and bacteria. However, only the rhinovirus is found more often during exacerbations; that is to say, pathogenic bacteria, mycoplasmas, and viruses other than rhinovirus are found just as often between as during exacerbations. Epidemiologic studies, however, implicate acute respiratory illness as one of the major factors associated with the etiology as well as the progression of chronic airways obstruction. Cigarette smokers may either transitorily develop or worsen small-airways obstruction in association with even mild viral respiratory infections. There is also some evidence that severe viral pneumonia early in life may lead to chronic obstruction, predominantly in small airways.
Genetics
Despite the strong etiologic association between smoking and COPD, only 15 to 20% of smokers lose FEV1 at a rate fast enough to manifest COPD. Epidemiologic evidence of familial clustering of COPD cases is strong and repeated, suggesting that susceptibility to the effects of tobacco smoke has genetic determinants. Twin studies show that even after controlling for active and passive smoking, FEV1 correlated more closely in monozygotic than dizygotic twins and more than in other family members with a lesser percentage of shared genotype. In first-degree relatives of a cohort of COPD patients with normal a 1AT levels, FEV1 was reduced compared to controls but only among current or ex-smokers. Smoking and nonsmoking relatives of control subjects both had normal FEV1. These data suggest genetic risk factors that are expressed in response to smoking.
a 1 Antitrypsin Deficiency
Thus far, deficiency of a 1AT is the only genetic abnormality that has been specifically linked to COPD. a 1AT is a 394-amino acid serine proteinase inhibitor whose synthesis is governed by a 12.2-kB 7-exon gene located at 14q32.1. a 1AT synthesis is expressed primarily in the liver and to a lesser degree in neutrophils and monocytes. Hepatic a 1AT escapes into the general circulation, where it counteracts neutrophil elastase. Normal levels of a 1AT are 20 to 48 m mol/L; levels above 11 mol/L (35% of normal) are considered protective. There are 75 known alleles of a 1AT, which are inherited in an autosomal codominant manner and are generally classified as normal (MM), deficient, null, or dysfunctional. The most common deficient allele, termed ZZ (or PiZZ phenotype), results from a single amino acid substitution 342Glu Lys, which causes spontaneous polymerization of the polypeptide, markedly impeding its release into the circulation from the liver. What does escape is vulnerable to oxidation and spontaneous polymerization, further impeding its function. The retained material is associated with hepatic cirrhosis (Chap. 299), while diminished circulating levels (2.5 to 7 mol/L, averaging 16% of normal) lead to antiprotease deficiency. PiZZ, the most common disease-related a 1AT abnormality, occurs in 1:2000 to 1:7000 persons of European descent and is rare in those of Oriental and African lineage. PiSS phenotypes are associated with a 1AT levels of 15 to 33 mol (mean 52% of normal). Pinull have no detectable antiprotease levels. Heterozygotes have intermediate levels of antiprotease.
Clinically significant deficiency of a 1AT, with levels below 11 mol/L, has been associated with homozygous PiZZ, Pinullnull, or PinullZ and the premature development of severe emphysema, chronic bronchitis, or bronchiectasis. a 1AT deficiency accounts for 2% of observed cases of emphysema. Rare below age 25, the disease usually presents as dyspnea and cough in patients in their fourth decade. Although not a true population-based study, a large national registry of 1129 severe a 1AT-deficiency cases indicated that the typical patient was in the mid-forties, with an FEV1 and a pulmonary diffusing capacity at or below 50% of the predicted levels. Most had exertional dyspnea and wheezing, but fewer than half reported a chronic cough. Nearly 80% had a positive family history of lung disease, and 25% reported a positive family history for liver disease. The average rate of decline of FEV1 is reported to be 100 to 130 mL per year for smokers and 50 to 80 mL per year for ex-smokers or lifetime nonsmokers with a 1AT deficiency.
Pathologically, panacinar emphysema predominates, and radiographically, changes are more marked in the lower lobes. It is becoming increasingly apparent that tobacco smoking is an extremely important cofactor for the development of disease in a 1AT-deficient individuals. Only a few lifetime nonsmokers with PiZZ develop emphysema. Most never have symptoms, have a normal rate of decline of FEV1, and live a normal life span. Many cases are discovered only as a consequence of family screening of emphysema patients. Because the total number of PiZZ individuals is unknown, the risk of disease for smokers is difficult to ascertain accurately. The risk of disease is lower still for heterozygotes with one M or S allele. Smoking is again an important cofactor.
Pathology
The pathologic changes of COPD involve large and small airways and the terminal respiratory unit. Airway narrowing is seen in large and small airways and is caused by changes in their normal constituents in response to persistent inflammation.
The airway epithelium is characterized by squamous metaplasia, atrophy of ciliated cells, and hypertrophy of mucus glands. The remodeled epithelium actively produces cytokines that amplify and sustain the inflammatory process. The small airways are the major site of airflow limitation. Small airways show a variety of lesions narrowing their lumina, including goblet cell hyperplasia, mucosal and submucosal inflammatory cells, edema, peribronchial fibrosis, intraluminal mucus plugs, and increased smooth muscle. CD8+ T lymphocytes and B lymphocytes characterize the inflammatory infiltrate. The marked thickening of the subepithelial lamina reticularis, characteristic of asthma, is absent in COPD.
In the central airways, subepithelial inflammation is present with increased numbers of eosinophils and CD8+ T lymphocytes. Unlike asthma, the eosinophils are not activated and do not degranulate. Neutrophils are present in the epithelium but not in the subepithelial layers. In larger cartilaginous airways, chronic bronchitis is associated with hypertrophy of submucosal mucus-producing glands. Quantitation of this anatomic change, known as the Reid index, is based on the ratio of the thickness of the submucosal glands to that of the bronchial wall. In persons without a history of chronic bronchitis, the mean ratio is 0.44 ± 0.09, whereas in those with such a history, the mean ratio is 0.52 ± 0.08. Although a low index is rarely associated with symptoms and a high index is commonly associated with symptoms during life, there is a great deal of overlap. Therefore, many persons will have morphologic changes in large airways without having had chronic bronchitis.
Emphysema begins as an increase in the number and size of alveolar fenestrae and results in the eventual destruction of alveolar septae and their attachments to terminal and respiratory bronchioles. Emphysema is classified according to the pattern of involvement of the gas-exchanging units (acini) of the lung distal to the terminal bronchiole. With centriacinar emphysema, the distention and destruction are mainly limited to the respiratory bronchioles with relatively less change peripherally in the acinus. Because of the large functional reserve in the lung, many units must be involved in order for overall dysfunction to be detectable. The centrally destroyed regions of the acinus have a high ventilation/perfusion ratio because the capillaries are missing, yet ventilation continues. This results in a deficit of perfusion relative to ventilation, while the peripheral portions of the acinus have crowded and small alveoli with intact, perfused capillaries giving a low ventilation/perfusion ratio. This results in a deficit of ventilation relative to blood flow, giving a high alveolar-arterial PO2 difference (PAO2 - PaO2) (Chap. 250).
During normal aging, airspaces enlarge and alveolar ducts increase in diameter. These changes are extremely common in lungs from persons over age 50 and may be misidentified as emphysema.
Panacinar emphysema involves both the central and peripheral portions of the acinus, which results, if the process is extensive, in a reduction of the alveolar-capillary gas exchange surface and loss of elastic recoil properties. When emphysema is severe, it may be difficult to distinguish between the two types, which most often coexist in the same lung.
Pathophysiology
Airflow Limitation
Although both chronic bronchitis and emphysema can exist without evidence of obstruction, by the time a patient begins to experience dyspnea as a result of these processes, obstruction is always demonstrable. Airflow limitation and increased airways resistance may be caused by loss of elastic recoil driving passive exhalation due to emphysema, by increased collapsibility of small airways through loss of radial traction on airways, or to increased resistance due to intrinsic narrowing of small airways.
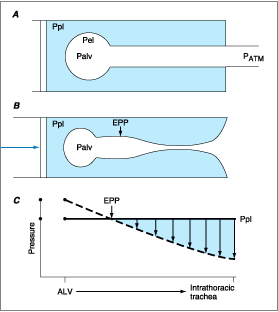
In addition to providing radial support to airways during quiet breathing, the elastic recoil properties of the lung serve as a major determinant of maximal expiratory flow rates. The static recoil pressure of the lung is the difference between alveolar and intrapleural pressure. During forced exhalations, when alveolar and intrapleural pressures are high, there are points in the airway at which bronchial pressure equals pleural pressure. Flow does not increase with higher pleural pressure after these points become fixed, so that the effective driving pressure between alveoli and such points is the elastic recoil pressure of the lung (Fig. 258-1). Hence maximal expiratory flow rates represent a complex and dynamic interplay among airways caliber, elastic recoil pressures, and collapsibility of airways. Correlative studies of structure and function suggest that small-airway narrowing is the most important correlate of airflow obstruction, followed by loss of elastic recoil. Collapsibility is probably a less important factor. As a direct consequence of the altered pressure-airflow relationships, the work of breathing is increased in bronchitis and emphysema. Since flow-resistive work is flow rate-dependent, there is a disproportionate increase in the work of breathing when ventilation must be increased, as in exercise.
Figure 258-1: A. A schematic diagram of the lung and intrathoracic airways with no airflow. The alveolar pressure (Palv) is greater than pleural pressure (Ppl) by an amount equal to the elastic recoil pressure of the lung (Pel)-i.e., Palv is the algebraic sum of Ppl + Pel. With no airflow Palv equals atmospheric pressure (PATM), and for all of the intrathoracic airways, pressure outside is less than the pressure inside due to the Pel. B. The same schematic lung during forced exhalation when pleural pressure becomes quite positive (arrow). Palv is still greater than Ppl by an amount equal to Pel. However, there is a pressure drop along the airway associated with flow, and at some point Ppl equals local bronchial pressure (so-called equal pressure point, EPP). Mouthward from this point, Ppl exceeds local bronchial pressure and hence acts to compress the airways. C. Pressure within the airways from alveoli to the intrathoracic trachea is shown as a dashed line (---) and Ppl is shown as a constant (-). Therefore, the driving pressure from alveoli to EPP is equal to Pel, and a decrease in Pel (i.e., loss of elastic recoil) would mean a smaller driving pressure and smaller flow rates.
Hyperinflation
The designated subdivisions of the lung volume outlined in Chap. 250 are abnormal to varying degrees in both bronchitis and emphysema. The residual volume and functional residual capacity (FRC) are almost always higher than normal. Since the normal FRC is the volume at which the inward recoil of the lung is balanced by the outward recoil of the chest wall, loss of elastic recoil of the lung results in a higher FRC. In addition, prolongation of expiration in association with obstruction would lead to a dynamic increase in FRC (dynamic hyperinflation) if inspiration is initiated before the respiratory system reaches its static balance point. Dynamic hyperinflation contributes additionally to the discomfort associated with airflow obstruction by flattening the diaphragm and placing it at a mechanical disadvantage due to shortened diaphragmatic fiber length and a perpendicular insertion with the lower ribs. The exertional increase in end-expiratory lung volume and consequent decrease in inspiratory capacity have been strongly associated with the degree of dyspnea. Elevations of total lung capacity (TLC) are frequent. The exact cause is uncertain, but increases in total lung capacity are often found in association with decreases in the elastic recoil of the lung. Although the vital capacity is frequently reduced, significant airways obstruction can be present with a normal to near-normal vital capacity.
Impaired Gas Exchange
Maldistribution of inspired gas and blood flow is always present to some extent. When the mismatching is severe, impairment of gas exchange is reflected in abnormalities of arterial blood gases. Small-airway narrowing causes a decrease in ventilation of their distal alveolar acini. When alveolar capillaries remain intact, this results in mismatching of ventilation and blood flow, reduced ventilation-perfusion ratios, and mild to moderate hypoxemia. With emphysema, destruction of alveolar walls may decrease alveolar capillary perfusion as well, better preserving ventilation-perfusion matching, and PaO2. Shunt hypoxemia is unusual. There are regions of the lung with a deficit of perfusion in relation to ventilation that increase the wasted ventilation ratio (i.e., Vd/Vt; Chap. 250). At a normal resting CO2 production, the net effective alveolar ventilation, as reflected by the arterial PCO2, may be excessive, normal, or insufficient, depending on the relationship of the overall minute volume to the wasted ventilation ratio.
The severity of gas exchange disturbances and, in large part, the clinical manifestations depend on the ventilatory response to the disordered lung function. Some patients, at the cost of extremely high effort of breathing and chronic dyspnea, maintain a strikingly increased minute volume, which results both in a normal to low arterial PCO2, despite the high Vd/Vt, and a relatively high arterial PO2, despite the high difference, PAO2 - PaO2. Other patients with only modest increases in effort of breathing and less dyspnea maintain a normal to only moderately elevated minute volume at the cost of accepting a high arterial PCO2 and a severely depressed arterial PO2.
Factors that account for clear differences in ventilatory responses among patients have been studied and debated for years. The bulk of available evidence suggests that those patients who maintain relatively normal or low arterial PCO2 levels are those with an increased ventilatory drive relative to their blood gas values, and those who chronically maintain high arterial PCO2 and lower PO2 levels have a diminished ventilatory drive in relation to their more severely deranged blood gas values. It is not at all certain whether individual differences are accounted for by variations in peripheral or central chemoreceptor sensitivity or through other afferent pathways.
Pulmonary Circulation
The pulmonary circulation malfunctions not only in terms of regional distribution of blood flow but also in terms of abnormal overall pressure-flow relationships. In advanced disease, there is often mild to severe pulmonary hypertension at rest, with further increases disproportionate to cardiac output elevations during exercise. A reduction in the total cross-sectional area of the pulmonary vascular bed can be attributed to thickening of medium and large muscular pulmonary arteries, to enhanced contraction of vascular smooth muscle in pulmonary arteries and arterioles, as well as to destruction of alveolar septa with loss of capillaries. Rarely does loss of capillaries alone lead to severe pulmonary hypertension with cor pulmonale, except as a near-terminal event (Chap. 237). Of more importance is the constriction of pulmonary vessels in response to alveolar hypoxia. The pulmonary arteries of patients with severe hypoxemia COPD have been shown to exhibit increased contractility and impaired relaxation in response to pharmacologic stimuli in vitro. These differences between the pulmonary arteries of COPD patients and normal individuals are abolished by inhibition of NO synthase, suggesting that patients develop an endothelial defect in NO synthesis. The constriction is somewhat reversible by an increase in alveolar PO2 with therapy.
There is a synergism between hypoxia and acidosis that assumes importance during episodes of acute or chronic respiratory insufficiency. Chronic hypoxia, especially in concert with carboxyhemoglobinemia, often seen with heavy cigarette smoking, leads not only to pulmonary vascular constriction but also to secondary erythrocytosis. The latter, although not proved to be a significant contributor to pulmonary hypertension, could add to pulmonary vascular resistance. As discussed in Chap. 237, chronic afterload on the right ventricle leads to hypertrophy and, in association with disordered blood gases, ultimately to failure. Hypoventilation may occur during rapid eye movement sleep and lead to desaturation, which may be severe. Repeated desaturation may cause pulmonary hypertension.
Renal and Hormonal Dysfunction
Chronic hypoxemia and hypercapnia have been shown to cause increased circulating levels of norepinephrine, renin, and aldosterone and decreased levels of antidiuretic hormone. Renal arterial endothelium in COPD patients exhibits defects similar to those seen in the pulmonary arteries, shifting renal blood flow from the cortex to the medulla and impairing renal functional reserve. The combination of hemodynamic and hormonal disturbances leads to defective excretion of salt and water loads and, together with right ventricular dysfunction, to the plethoric and cyanotic manifestations of some patients with COPD.
Cachexia
Weight loss sometimes occurs in patients with advanced COPD. A body-mass index (BMI) < 25 kg/m2 is associated with increased frequency of exacerbations and with significantly reduced survival. Cachexia has been attributed to caloric intake failing to keep pace with energy expenditures associated with increased work of breathing, but more recent evidence suggests that a biochemical basis is more likely. Hypoxemia leads to increased circulating levels of tumor necrosis factor (TNF) and weight loss has now been correlated with levels of the latter.
Peripheral Muscle Dysfunction
Protein and muscle are lost as part of wasting in advanced COPD. Skeletal muscle bulk is lost with proportional reductions in strength. Proximal limb girdle muscles of the upper and lower extremities are particularly affected, contributing to dyspnea with activities of daily living. Fiber composition in skeletal muscle changes, favoring endurance over strength. These changes occur in parallel with FEV1 and independently of glucocorticoid use, which can also cause myopathy and muscle weakness.
Osteoporosis
Loss of bone density is common in advanced disease. Over half of COPD patients lose more than 1 SD of bony density, and more than one-third have values more than 2 SDs below normal. Vertebral fractures are especially common. These changes are even more severe in patients receiving chronic glucocorticoid therapy.
Natural History
COPD is identified by the presence of an abnormal FEV1 in middle age, usually early in the fifth decade, and is characterized by an accelerated decline of FEV1 with aging. In normal individuals, FEV1 normally reaches a lifetime peak at age 25 and undergoes a linear decline of about 35 mL per year thereafter. Annual loss of FEV1 among susceptible individuals who develop COPD is between 50 and 100 mL per year. Greater rates of decline have been associated with mucus hypersecretion, especially in men, and with bronchial hyperreactivity. Acute exacerbations do not alter the rate of decline. Dyspnea and impairment of physical work capacity are characteristic only of moderately severe to severe airways obstruction. There is considerable variation among individual patients. The majority of patients usually experience exertional dyspnea when FEV1 falls below 40% of predicted and have dyspnea at rest when the FEV1 <25% of predicted. In addition to dyspnea at rest, CO2 retention and cor pulmonale frequently occur when the FEV1 falls to 25% of predicted. With a respiratory infection, small changes in the degree of obstruction can make a large difference in symptoms and gas exchange. Thus small therapeutic gains may have rewarding results.
Exacerbation
The clinical course of COPD can be characterized as one of slow progression and relative stability punctuated by episodic exacerbations occurring, on average, a little more than once per year. Exacerbations are generally described as a worsening of previously stable disease characterized by increased dyspnea, wheeze, and cough and sputum volume, tenacity, and purulence, with variable degrees of water retention and with worsening gas exchange and ventilation-perfusion relationships. Hyperinflation and work of breathing are increased. To the extent that diaphragmatic function and neuromuscular drive can compensate for the increased work, PaCO2 will not rise, but when work demands exceed respiratory pump capacity, hypercapnia and respiratory acidemia ensue. Cardiac output often does not increase sufficiently to compensate for the increased oxygen consumption from respiratory muscles, thereby compounding the hypoxemia due to V/Q mismatching and hypercapnia.
Most COPD exacerbations are thought to be a consequence of acute tracheobronchitis, usually infectious. Most infections are primarily bacterial or the consequence of bacterial superinfection of a primary viral process. Exacerbations may also be triggered by, and must be distinguished from, left ventricular failure, cardiac arrhythmias, pneumothorax, pneumonia, and pulmonary thromboembolism. Upper airway obstruction, aspiration, rhinitis or sinusitis, asthma, or gastroesophageal reflux should be excluded. Although COPD exacerbations are individually serious and potentially life-threatening, they do not cause accelerated declines of FEV1 over time.
Clinical Manifestations
History
Patients with COPD are most often tobacco smokers with a history of at least one pack per day for at least 20 years. The disease is only rarely seen in nonsmokers. Onset is typically in the fifth decade and often comes to attention as a productive cough or acute chest illness. Exertional dyspnea is usually not encountered until the sixth or seventh decade. The patient's perception of dyspnea correlates poorly with physiologic measurements, especially among older patients. A morning "smoker's cough" is frequent, usually mucoid in character but becoming purulent during exacerbations, which in early disease are intermittent and infrequent. Volume is generally small. Production of more than 60 mL/d should prompt investigation for bronchiectasis. The frequency and severity of cough generally do not correlate with the degree of functional impairment. Wheezing may be present but does not indicate severity of illness. As COPD progresses, exacerbations become more severe and more frequent. Gas exchange disturbances, worsen and dyspnea becomes progressive. Exercise tolerance becomes progressively limited. With worsening hypoxemia, erythrocytosis and cyanosis may occur. The development of morning headache may indicate the onset of significant CO2 retention. In advanced disease, weight loss is frequent and correlates with an adverse prognosis. When blood gas derangements are severe, cor pulmonale may manifest itself by peripheral edema and water retention. Anxiety, depression, and sleep disturbances are not infrequent.
The physical examination has poor sensitivity and variable reproducibility in COPD. Findings may be minimal or even normal in mild disease, requiring objective laboratory data for confirmation. In early disease, the only abnormal findings may be wheezes on forced expiration and a forced expiratory time prolonged beyond 6 s. With progressive disease, findings of hyperinflation become more apparent. These include an increased anteroposterior diameter of the chest, inspiratory retraction of the lower rib margins (Hoover's sign), decreased cardiac dullness, and distant heart and breath sounds. Coarse inspiratory crackles and rhonchi may be heard, especially at the bases. To gain better mechanical advantage for their compromised respiratory muscles, patients with severe airflow obstruction may adopt a characteristic tripod sitting posture with the neck angled forward and the upper torso supported on the elbows and arms. Breathing through pursed lips prolongs expiratory time and may help reduce dynamic hyperinflation.
Cor pulmonale and right heart failure may be evidenced by dependent edema and an enlarged, tender liver (Chap. 237). With pulmonary hypertension, a loud pulmonic component of the second heart sound may be audible, along with a right ventricular heave and a murmur of tricuspid regurgitation; these findings may be obscured by hyperinflation. If right-sided pressures are sufficiently high, neck veins may elevate instead of collapse with inspiration (Kussmaul's sign). Cyanosis is a somewhat unreliable manifestation of severe hypoxemia and is seen when severe hypoxemia and erythrocytosis are present.
Radiographic Findings
A posteroanterior and lateral chest film should be obtained primarily to exclude competing diagnoses. They may be entirely normal in mild disease. As COPD progresses, abnormalities reflect emphysema, hyperinflation, and pulmonary hypertension. Emphysema is manifested by an increased lucency of the lungs. In smokers, these changes are more prominent in the upper lobes, while in 1AT deficiency, they are more likely in basal zones. Local radiolucencies >1 cm in diameter and surrounded by hairline arcuate shadows indicate the presence of bullae and are highly specific for emphysema. With hyperinflation, the chest becomes vertically elongated with low flattened diaphragms. The heart shadow is also vertical and narrow. The retrosternal airspace is increased on the lateral view, and the sternal-diaphragmatic angle exceeds 90°. In the presence of pulmonary hypertension, the pulmonary arteries become enlarged and taper rapidly. The right heart border may become prominent and impinge on the retrosternal airspace. The presence of "dirty lung fields" may reflect the presence of bronchiolitis.
Computed tomography has greater sensitivity and specificity for emphysema than the plain film but is rarely necessary except for the diagnosis of bronchiectasis and evaluation of bullous disease. Nonhomogeneous distribution of emphysema is thought by some to be an indicator of suitability for lung volume reduction surgery (LVRS).
Pulmonary Function Testing
Because of the imprecision of clinical findings, objective evaluation of the presence, severity, and reversibility of airflow obstruction is essential in the diagnostic evaluation of COPD. A normal FEV1 essentially excludes the diagnosis. The spirogram in COPD shows decreased volume changes with time and a failure to reach a plateau after 3 to 5 s. Continued airflow may be evident for 10 s or more on forced exhalation. The flow-volume curve shows diminished expiratory flow at all lung volumes. Expiratory flow is concave to the volume axis. When flow is plotted against absolute lung volume, the entire curve is shifted to higher volumes, reflecting hyperinflation. Serial spirometry is important in assessing the rate of decline of FEV1.
Reversibility is assessed by spirometry before and after administration of an inhaled bronchodilator, most often a short-acting b 2-adrenergic agonist. Testing should be performed when the patient is clinically stable. Short-acting bronchodilators should be withheld for 6 h, long-acting dilators for 12 h, and theophylline for 24 h prior to testing. A significant response is an increase of at least 12% and 200 mL in either FEV1 or forced vital capacity (FVC). Postbronchodilator FEV1 is useful for prognostication. Although only one-third of COPD patients show a significant response to an inhaled bronchodilator in the pulmonary function laboratory on any one day, two-thirds will show a significant response when tested with different bronchodilators on several different occasions. The degree of bronchodilator response at any one testing session does not predict the degree of clinical benefit to the patient. Therefore, bronchodilators are given irrespective of the acute response obtained in the pulmonary function laboratory. The American Thoracic Society recommends staging COPD by FEV1. Stage I, mild disease, is defined as FEV1 >50% predicted; stage II, moderate disease, 35 to 49% predicted; and stage III, severe disease, <35% predicted.
Lung volumes are useful for the assessment of hyperinflation. Transfer factor for carbon monoxide (DLCO) correlates negatively with the degree of emphysema but is not specific and may miss mild disease. Neither test is indicated routinely, but DLCO may help distinguish chronic asthma from emphysema.
Measurements for arterial blood gas are not needed for mild disease, but they should be assessed routinely for stage II or stage III COPD. Patients with pulmonary hypertension or cor pulmonale with normal daytime blood gases should be evaluated for nocturnal desaturation by overnight oximetry. Polysomnography to exclude concurrent sleep apnea should be obtained for patients who also complain of excessive daytime somnolence or who have a history of snoring.
a
1AT levels are not needed routinely but should be obtained for chronic airflow obstruction or chronic bronchitis in nonsmokers, as well as in COPD patients with bronchiectasis, cirrhosis without apparent risks, premature emphysema, or basilar emphysema; in patients under age 50 with unremitting asthma; and in individuals with a family history of a 1AT deficiency.Treatment
Treatment of COPD is based on the principles of prevention of further evolution of disease, preservation of airflow, preservation and enhancement of functional capacity, management of physiologic complications, and avoidance of exacerbations.
Smoking Cessation
The Lung Health Study has demonstrated that elimination of tobacco smoking confers significant survival benefit to patients with COPD. Prolonged survival is associated with reduced rates of malignancy and cardiovascular disease as well as with a significant increment in FEV1 in the first year after smoking cessation. The rate of decline of FEV1 reverts back to that of a nonsmoker. Although bronchodilator therapy produces similar first-year gains in FEV1, pharmacotherapy alone does not modify the decline of airflow over time. Even unsuccessful quitters show significant benefits when compared to continuing smokers.
Despite the demonstrated benefits of smoking cessation, sustained quitting is difficult to achieve. Overall, only 6% of smokers succeed in quitting long term, and 70 to 80% of short-term quitters start smoking again. Successful quitting requires concerted active and continuing intervention by the physician. The physician should address the issue in regular patient visits, assess the patient's readiness to quit, advise the patient as to the best methods for smoking cessation, provide emotional and pharmacologic support, and arrange close follow-up of the patient's efforts. The concept of "lung age" may be helpful in promoting smoking cessation by determining the age at which the observed FEV1 would be a normal finding. Lungs of 50- to 60-year-old smokers may be "normal" for a 70- to 80-year-old individual. Nicotine patches and nicotine polacrilex gum improve quit rates, especially among nicotine-dependent smokers. The addition of oral bupropion at 150 mg twice daily produces significant additional benefit, with a 1-year sustained abstinence rate of 22.5% compared to 6% for placebo. Smoking cessation is typically associated with weight gain of 3 to 4 kg. To minimize weight gain, reluctance to quit, and relapse, prospective quitters should be counseled to reduce caloric intake and to increase physical activity.
Bronchodilators
These drugs improve dyspnea and exercise tolerance by improving airflow and by reducing end-expiratory lung volume and air-trapping. Although airflow limitation is relatively fixed, some degree of response to bronchodilator medication is usually present. Bronchodilator medication is available in metered-dose inhaler (and some dry-powder inhalers) and in nebulizable and oral forms. Inhalers deliver medications directly to the airways and have limited systemic absorption and side effects. Proper use requires timing and coordination of inspiration and inhaler actuation and presents frequent difficulties for chronic lung patients. These problems can usually be overcome with education and with the use of holding chambers. Aerosol nebulizers have no pharmacologic advantage over metered-dose inhalers. Their use should be limited to patients who remain unable to master metered dose inhalers adequately. Oral medication is associated with higher rates of adherence than inhalers but shows higher rates of systemic side effects without superior bronchodilation.
Three major classes of bronchodilators are commonly employed in the treatment of patients with COPD: short- and long-acting b 2-adrenergic agonists, anticholinergics, and theophylline derivatives. Short-acting b 2-agonists (albuterol, pirbuterol, terbutaline, metaproterenol) are relatively bronchoselective with minimal effects on heart rate and blood pressure. They produce significant bronchodilation at 5 to 15 min and remain effective for 4 to 6 h. Long-acting b 2-agonists (oral sustained-release albuterol and inhaled salmeterol) have an onset of action of 15 to 30 min and a 12-h duration of action. Anticholinergic agents (ipratropium bromide) have a 30- to 60-min onset of action and a 4- to 6-h duration. Theophyllines are generally administered orally in 12- or 24-h preparations. Recommended bronchodilator regimens are shown in Table 258-1.
Table 258-1: Recommended Bronchodilator Therapy for Chronic Obstructive Pulmonary Disease
|
Stage |
% predicted |
Treatment |
||||||||
|
I |
> 50 |
b 2-agonist prn |
||||||||
|
II |
35-49 |
Combined anticholinergic and b 2-agonist |
||||||||
|
III |
<35 |
Above plus long-acting b2-agonist and/or sustained release theophylline |
||||||||
Regular use of ipratropium may lead to improvements in baseline FEV1 when compared with short-acting b 2-agonists. When used together, ipratropium and short-acting b 2-agents show greater clinical efficacy than either agent alone, without an increase in side effects. Salmeterol as a single agent produces longer lasting bronchodilation than ipratropium, improves baseline FEV1 over time, and is not associated with loss of efficacy over a period of several months. Salmeterol, however, has not yet been evaluated as a component of combination therapy.
Theophylline is a weak bronchodilator with a narrow therapeutic window. Much of its clinical benefit derives from effects other than bronchodilation; therapeutic doses of theophylline increase ventilatory drive, enhance diaphragmatic contractility, and increase cardiac output. About 20% of COPD patients respond to theophylline with improved airflow, exercise tolerance, and quality of life. Theophylline produces additional benefits in exercise capacity and quality of life when used in combination with short-acting b 2-adrenergic agonists. The therapeutic range for theophylline is commonly given as 10 to 20 m g/mL, with greater efficacy but greater toxicity seen at higher serum levels. The risk of toxicity is greater in older patients and in those with heart and kidney disease. Optimal dosing must balance the competing considerations of risk and benefit for each individual patient.
Glucocorticoids
Because COPD, like asthma, is a disease associated with airway inflammation, glucocorticoids are an intuitively attractive therapeutic modality. Nevertheless, results of clinical trials of glucocorticoid therapy in COPD patients have shown less impressive benefits when compared to patients with asthma. The degree of response to glucocorticoids appears to correlate with the presence of asthmatic features, but data supporting their use is limited. Only 10% more patients show subjective benefit and increase their FEV1 or forced vital capacity by at least 20% when compared to those on placebo. Responders cannot be reliably identified on clinical grounds, although response to an inhaled b 2-agonist is commonly used as a predictor. The benefits of a 10- to 14-day trial of 30 to 40 mg/d of prednisone for patients with stage III disease who have not responded adequately to mixed bronchodilator therapy remain to be proven. Long-term systemic glucocorticoid use is associated with multiple side effects. In particular, they have been associated with worsened osteoporosis and increased risk of vertebral fracture. If systemic steroids are used, the lowest effective dose should be employed and alternate-day dosing used whenever possible. The use of inhaled glucocorticoids ameliorates systemic side effects. Three large clinical trials have shown that inhaled glucocorticoids do not alter the rate of decline of FEV1. While an inhaled glucocorticoid does not decrease the number or frequency of COPD exacerbations, it may decrease their severity and reduce the need for hospitalization. Symptoms and exercise tolerance improve on inhaled glucocorticoids.
Management of a AT deficiency
Given the central role of smoking in the pathogenesis of disease, smoking cessation is an important cornerstone in the management of a 1AT deficiency. Exogenous a 1AT derived from pooled human plasma administered intravenously in a weekly dose of 60 mg/kg has been shown to induce protective levels of a 1AT in deficient individuals. Because of the expense and inconvenience of the treatment, replacement of a 1AT is used only for patients over age 18 with a 1AT levels below 11 m mol/L who have stopped smoking and who have airflow obstruction. A recently published large nonrandomized trial showed that augmentation therapy significantly decreased 5-year mortality (RR 0.64) for patients receiving replacement. The rate of decline of FEV1 also decreased with augmentation therapy. In both instances, benefit was largely restricted to those patients with FEV1 35 to 49% of predicted. These findings require confirmation in randomized controlled trials.
Oxygen
Severe and progressive hypoxemia is often seen in advanced COPD and may result in cellular hypoxia with deleterious physiologic consequences. The establishment of adequate systemic oxygen transport is essential to the prevention of tissue hypoxia and requires attention to cardiac output and hemoglobin concentration as well as to arterial O2 saturation (SaO2). Long-term O2 therapy has been shown to reverse secondary polycythemia; improve body weight; ameliorate cor pulmonale; and enhance neuropsychiatric function, exercise tolerance, and activities of daily living. Two major studies, one in the United States and one in the United Kingdom, established a survival benefit for long-term O2 therapy that increased with the number of hours per day that O2 was used. The mechanism for this benefit has not been conclusively elucidated, but it appears to be related to the stabilization of pulmonary hemodynamics.
The need for long-term O2 therapy should be documented with measurement of arterial blood gases obtained at rest and confirmed by a separate determination of resting arterial blood gases during a period of medical stability after 30 to 90 days of optimum medical therapy. Once the need for O2 has been demonstrated in a stable patient, the requirement is generally for the duration of the patient's life. Patients with a PaO2 >55 mmHg or SaO2 >88% should be provided with oxygen titrated to raise SaO2 to >90%. Oxygen is likewise indicated for patients who have a PaO2 of 56 to 59 mmHg with SaO2 >89% when hematocrit is >55% or when cor pulmonale or other objective evidence of pulmonary hypertension is present. Oxygen may be appropriate for patients whose resting awake PaO2 >60 mmHg with SaO2 >90% if they become hypoxic during exercise or sleep. Once oxygen is prescribed, the dose should be titrated to maintain SaO2 >90% during sleep and normal walking, as well as at rest, and it should be used for a minimum of 15 h a day to realize a survival benefit.
Oxygen is most frequently delivered through a nasal cannula at rates of 2 to 5 L/min. Oxygen-sparing cannulae are available. Transtracheal administration provides further O2-sparing benefits but requires scrupulous attention to catheter maintenance and hygiene and is not suitable for all patients. Oxygen is packaged as compressed gas or compressed liquid or can be delivered from an O2concentrator, a molecular sieve that enriches O2 by removing nitrogen from ambient air. O2 should be prescribed from sources that are appropriate to the individual patient's life-style and needs. It is customary to provide a stationary O2 source, either an O2 concentrator, which is dependent on a reliable source of electricity, or 100-kg (200-lb) H cylinders of compressed O2. Flow resistance imposes a 15-m (50-ft) practical limit to the length of tubing connecting the O2 source to the patient's cannula. For patients whose activities of daily living require ambulation beyond this limit, ambulatory or portable systems should be provided. Ambulatory O2 needs may be met with rolling 10-kg (22-lb) E cylinders of compressed O2, or with portable 2-kg (4.5-lb) aluminum cylinders or 3-kg (6.6-lb) liquid oxygen packs. The duration of O2 availability from an O2 concentrator is unlimited. For compressed gas and liquid sources, the amount of available oxygen is determined by the size of the system and the patient's liter flow needs. Portable systems generally provide 4 to 5 h of O2 flow.
Oxygen therapy is generally safe. Cylinders should be secured to prevent tipping over or potentially explosive disconnection of the regulator valve. Oxygen should be stored away from open flames or other source of heat, and patients and family members should be educated to be especially scrupulous about avoiding smoking in the presence of flowing O2.
Prophylaxis
No evidence supports the prophylactic use of antibiotics in stable COPD. Yearly influenza vaccination is recommended for all patients with chronic cardiopulmonary disease, although objective benefit has not been conclusively demonstrated. Pneumococcal vaccination with 23-valent polysaccharide is also recommended. Amantadine should be used for unvaccinated patients who are placed at risk by an outbreak of influenza A.
Rehabilitation
Airflow limitation, dyspnea, and muscle loss and deconditioning all compromise cardiopulmonary fitness and contribute to a progressively constrained daily life and unsatisfactory quality of life. Pulmonary rehabilitation is a multidisciplinary program of care for patients with chronic respiratory impairments that is individually tailored and designed to optimize physical and social performance. A pulmonary rehabilitation program consists of exercise training, patient education, psychosocial and behavioral intervention, and regular assessment of outcomes and is designed to minimize the disability and handicap imposed by the physiologic impairments consequent to COPD. Rehabilitation in COPD should be considered for patients with persistent symptoms and disability despite optimal medical management. Spirometric criteria should not be the primary basis for referral into rehabilitation programs. Exercise consists of 20 to 30 min of upper and lower extremity exercise at 60 to 75% maximum VO2 or heart rate two to five times a week. Both strength and endurance exercises are provided. Education covers pursed lip and other breathing strategies to minimize dyspnea, energy-conservation skills, principles of medications and proper use of metered-dose inhalers, nutrition, and end-of-life decision-making. Behavioral interventions focus on dyspnea, depression, and self-sufficiency and on issues of control, coping, and role function. Dyspnea, exercise tolerance, activity level, and quality of life are followed at regular intervals. Pulmonary rehabilitation programs have been shown to improve endurance time for submaximal exercise by 38 to 80% and 6-min walking distance by 80 to 113 m. Clinically meaningful reduction in dyspnea and improvement of quality of life have been reported. No clinical trials have been adequately designed to address the issue of survival benefit. Reductions in costs of care and resource consumption have not reached statistical significance.
Despite maximal medical therapy, when COPD progresses to stage III and is complicated by hypercapnia or pulmonary hypertension, surgical approaches to treatment may be considered.
Transplantation
Owing to its frequency in the general population, emphysema is the most common indication for lung transplantation. Transplantation should be actively considered for end-stage COPD patients when the prognosis from the disease is worse than the survival statistics for the surgery. Lung transplantation should be considered for COPD patients who, despite maximal medical therapy, have an FEV1 < 25% predicted and with pulmonary hypertension or cor pulmonale. Precedence is given to those patients with a PaCO2 of 55 mmHg and progressive deterioration. Asthma and other reversible airflow limitation must be excluded. Rehabilitation and long-term O2 therapy, where appropriate, should be provided prior to transplant evaluation.
Lung Volume Reduction Surgery
LVRS, or pneumectomy, is designed to relieve dyspnea and improve exercise function in severely disabled patients with stage III emphysema. At operation, severely emphysematous lung tissue is resected, leading to improvement in elastic recoil in the remaining pulmonary parenchyma. This decreases hyperinflation and enhances diaphragmatic function, with consequent 25 to 50% improvement of airflow and exercise capacity. In early uncontrolled studies, hospital mortality for LVRS ranged from 5 to 18% and hospital stays averaged 9 to 18 days, with frequent significant air leaks. Cost of LVRS was $33,000 to $70,000 per case. Because of the large number of potential candidates, the high cost involved, and unanswered questions about the benefits of the operation, use of LVRS in the United States has been restricted to a multicenter randomized controlled trial, the National Emphysema Treatment Trial (NETT), comparing LVRS with best medical therapy. Stage III emphysema patients accepted for evaluation into NETT are under age 75, are severely hyperinflated, and have severe dyspnea despite optimal medical therapy. Contraindications to LVRS are similar to those for lung transplantation, including active smoking, marked obesity or cachexia, and inability to undertake pulmonary rehabilitation successfully. There has been little consensus regarding features identifying ideal and suboptimal candidates for the surgery. Radiographic heterogeneity of disease and the absence of significant intrinsic airway disease have been suggested characteristics of patients likely to benefit. Results from the NETT suggest that physiologic benefits from LVRS may begin to be lost as early as 1 year after surgery. Accelerated declines of FEV1 have been reported, averaging 100 mL per year and particularly marked in those patients with the greatest postoperative gains in airflow. Improvements in dyspnea and exercise tolerance may be sustained for as long as 3 years but may decline thereafter. Until these issues are satisfactorily resolved, LVRS will remain an experimental procedure.
Treatment of Exacerbations
Triage
The initial decision in the management of an exacerbation of COPD is whether hospitalization is necessary. Rapidity of evolution of symptoms and response to initial therapy, level of consciousness, presence or absence of respiratory distress, severity of gas exchange disturbance, and arterial blood gas deviation from the patient's stable baseline should influence the decision to hospitalize. The patient's ability to manage at home and the resources available for home care should weigh heavily in the decision-making process.
Home Therapy
For patients with mild exacerbations for whom outpatient therapy is appropriate, a combination of anticholinergic and short-acting b 2-adrenergic agonist bronchodilators should be prescribed. Although b 2-agonists may be given as frequently as once an hour, there is no advantage to administering anticholinergic bronchodilators more frequently than every 4 to 6 h. Metered-dose inhalers should be used with spacers. There is no evidence that the use of nebulizers provides any improvement in outcome.
The presence of increased sputum volume or purulence suggest an infectious cause of an exacerbation. With either of these features is present in conjunction with increased breathlessness or when both are present, antibiotics should be prescribed. The organisms most frequently associated with mild COPD exacerbations include Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis. Trimethoprim/sulfamethoxazole, doxycycline, or amoxicillin is an appropriate management option, although choices may be modified by local antibiotic sensitivity data.
There is a need for well-controlled studies on the utility of glucocorticoids in the outpatient management of COPD exacerbations. Oral glucocorticoids may be continued in patients already receiving such treatment or given to patients who do not show a satisfactory response to bronchodilator therapy. The usual dose is 20 to 40 mg daily for 7 to 10 days. Short-term glucocorticoid therapy lasting less than 3 weeks may be discontinued without the use of a tapering dose.
Hospital Management
For patients with exacerbations of sufficient severity to warrant hospitalization, improvement of airflow, gas exchange, and acid-base status are of central importance. Hospitalized patients should receive bronchodilators, antibiotics, oral glucocorticoids, and sufficient O2 to keep the SaO2 >90%. b 2-agonists and anticholinergic agents should be given together every 4 to 6 h. The frequency of sympathomimetic bronchodilator administration may be increased as needed to as often as every 20 min. Because high doses of b 2-agonists may cause hypokalemia, serum potassium levels and heart rate should be monitored closely for patients receiving frequent doses of these agents. Data are contradictory regarding the addition of theophylline to the bronchodilator regiment of patients showing an inadequate initial response, yet the current American and British Thoracic Societies' guidelines recommend consideration of its use to produce plasma theophylline levels between 10 and 20 m g/mL. Oral glucocorticoids have been shown to produce modest improvements in FEV1 and in the duration of hospitalization for COPD exacerbations. Recent data indicate that more severe COPD exacerbations are associated with the recovery of enterobacteriaciae in respiratory secretions. For this reason, a second- or third-generation cephalosporin, a fluoroquinolone, a second-generation macrolide, or an extended-spectrum penicillin is now recommended as initial therapy. Attempts to obtain diagnostically adequate sputum should be made, and, when available, sputum results should be used to individualize therapy in the light of local microbial sensitivity spectra. Oxygen therapy is an important component of the management of a severe exacerbation of COPD. It is important to maintain the SaO2 > 90% and PaO2 between 60 and 65 mmHg for most patients. In many cases, administration of O2 will result in worsening hypercapnia, although rarely to a clinically significant degree if the O2 is used only in amounts to achieve the minimal goals. The elevation of PaCO2 is multifactorial, resulting from increased dead space due to reduced tidal volume as well as from the Haldane effect, i.e., a right wave shift of the CO2 dissociation curve in the presence of increased saturated hemoglobin. The lower the initial PaO2 and the greater the increase, the larger the increase in PaCO2 observed. Patients whose pH on presentation is below 7.25 and with PaO2 < 50 mmHg are at particular risk and should be observed closely.
For patients at increased risk of hypercapnia, administration of controlled concentrations of O2 through a Venturi mask is reasonable. Inspired O2 concentrations (FIO2) of 0.24 to 0.28 are usually sufficient to keep SaO2 90%.
Mechanical Ventilation
Patients with impaired consciousness, respiratory distress evidenced by tachypnea with a respiratory rate greater than 35 breaths per minute and/or abdominal paradox, severe hypoxemia, or significant respiratory acidosis with pH < 7.25 and who deteriorate despite treatment are candidates for immediate ventilatory support using either noninvasive (mask) or invasive (intubation) approaches. The goals are to buy time for medical treatments to take effect, to rest the respiratory muscles, and to improve gas exchange abnormalities while avoiding the major complications of mechanical ventilatory support.
Noninvasive positive-pressure ventilation (NIPPV) delivered by nasal mask should be considered in units that have experience with the technique for patients who remain alert and cooperative, who are not heavily sedated, who are hemodynamically stable, and who are able to clear their airways by coughing up secretions. In these circumstances, NIPPV has been shown to be successful in avoiding the need for endotracheal intubation in up to 70% of cases. Success, as evidenced by improved PaCO2 and pH, should be evident within the first 60 min. Part-time NIPPV for 6 to 8 h per day may afford sufficient respiratory muscle rest to avert the need for invasive conventional ventilation. Failed attempts at NIPPV can be followed by intubation and conventional ventilation and do not appear to carry a worse prognosis. Successful application of NIPPV has been associated with a decrease in intensive care and hospital stays, incidence of nosocomial pneumonia, and costs.
Before committing to endotracheal intubation and conventional ventilatory support, the patient's wishes for such support, the patient's quality of life, and the benefits and costs of care should be thoroughly reviewed. Where the patient's wishes cannot be clearly ascertained or there is uncertainty about the appropriateness of the intervention, intubation and ventilation should proceed. If mechanical ventilatory support is subsequently determined to be inappropriate, support may then be withdrawn.
Once intubation is accomplished, the patient can be ventilated in the controlled ventilation, assist-control, intermittent mandatory ventilation, or pressure support modes. FIO2 should be sufficient to obtain SaO2 >90% and PaO2 of 60 to 65 mmHg. An FIO2 of 0.24 to 0.40 is usually adequate for the purpose. Minute volume should be adequate to keep pH >7.25, but one should not strive to achieve a "normal" PaCO2. It is important to try to avoid overventilation and hyperinflation in ventilated COPD patients. Because the time constant for exhalation is abnormally prolonged, it is essential to allow adequate expiratory time to permit as complete emptying of each breath as possible, preferably at least 3 to 4 s. This is best accomplished by minimizing tidal volume and respiratory rate. Lesser gains in expiratory (E) time can be obtained by high inspiratory (I) flow rates and I:E ratios of 1:2 or higher. Inadequate expiratory time leads to dynamic hyperinflation and in turn to the development of intrinsic positive end-expiratory pressure (PEEPi). PEEPi is just as capable of producing hypotension as extrinsically applied PEEP. When a mechanically ventilated patient with obstructive lung disease abruptly develops hypotension, PEEPi should be excluded, either by direct measurement or by disconnecting the patient from the ventilator for 30 to 60 s. PEEPi and dynamic hyperinflation increase the work of breathing, place the diaphragm at mechanical disadvantage, and contribute significantly to difficulties in weaning from ventilatory support. Over a period of days, as the underlying precipitants of the exacerbation are controlled, airway obstruction gradually remits and gas exchange improves and it becomes appropriate to consider removal from mechanical ventilatory support.
Prognosis after Exacerbation
The hospital mortality rate for an episode of respiratory failure in COPD ranges from 11 to 25% and depends on the severity of the episode, the patient's chronic health and nutritional status, and the presence of cor pulmonale or congestive heart failure. Data regarding subsequent course may be helpful in educating COPD patients and in guiding their subsequent management decisions. Among survivors of mechanical ventilation, the 6-month mortality rate is approximately 40%. Two-thirds of survivors have frequent recurrences of exacerbations, and functional status thereafter is often poor.
