Cirrhosis and Its Complications
Cirrhosis
is a pathologically defined entity that is associated with a spectrum of
characteristic clinical manifestations. The cardinal pathologic features
reflect irreversible chronic injury of the hepatic parenchyma and include
extensive fibrosis in association with the formation of regenerative nodules.
These features result from hepatocyte necrosis, collapse of the supporting
reticulin network with subsequent connective tissue deposition, distortion of
the vascular bed, and nodular regeneration of remaining liver parenchyma. The
central event leading to hepatic fibrosis is activation of the hepatic stellate
cell. Upon activation by factors released by hepatocytes and Kupffer cells, the
stellate cell assumes a myofibroblast-like conformation and, under the
influence of cytokines such as transforming growth factor ![]() (TGF-
(TGF-![]() ),
produces fibril-forming type I collagen. The precise point at which fibrosis
becomes irreversible is unclear. The pathologic process should be viewed as a
final common pathway of many types of chronic liver injury. Clinical features
of cirrhosis derive from the morphologic alterations and often reflect the
severity of hepatic damage rather than the etiology of the underlying liver disease.
Loss of functioning hepatocellular mass may lead to jaundice, edema,
coagulopathy, and a variety of metabolic abnormalities; fibrosis and distorted
vasculature lead to portal hypertension and its sequelae, including
gastroesophageal varices and splenomegaly. Ascites and hepatic encephalopathy
result from both hepatocellular insufficiency and portal hypertension.
),
produces fibril-forming type I collagen. The precise point at which fibrosis
becomes irreversible is unclear. The pathologic process should be viewed as a
final common pathway of many types of chronic liver injury. Clinical features
of cirrhosis derive from the morphologic alterations and often reflect the
severity of hepatic damage rather than the etiology of the underlying liver disease.
Loss of functioning hepatocellular mass may lead to jaundice, edema,
coagulopathy, and a variety of metabolic abnormalities; fibrosis and distorted
vasculature lead to portal hypertension and its sequelae, including
gastroesophageal varices and splenomegaly. Ascites and hepatic encephalopathy
result from both hepatocellular insufficiency and portal hypertension.
Classification
of the various types of cirrhosis based on either etiology or morphology alone
is unsatisfactory. A single pathologic pattern may result from a variety of
insults, while the same insult may produce several morphologic patterns.
Nevertheless, most types of cirrhosis may be usefully classified by a mixture
of etiologically and morphologically defined entities as follows: (1) alcoholic;
(2) cryptogenic and posthepatitic; (3) biliary; (4) cardiac; and (5) metabolic,
inherited, and drug-related. This chapter considers the various types of
cirrhosis and their complications.
Alcoholic
Cirrhosis
Definition
Alcoholic cirrhosis is only one of many consequences resulting from
chronic alcohol ingestion, and it often accompanies other forms of
alcohol-induced liver injury, including alcoholic fatty liver and alcoholic
hepatitis (Chap. 298). Alcoholic cirrhosis, historically referred to as Laennec's
cirrhosis, is the most common type of cirrhosis encountered in North
America and many parts of western Europe and South America. It is characterized
by diffuse fine scarring, fairly uniform loss of liver cells, and small
regenerative nodules, and therefore it is sometimes referred to as micronodular
cirrhosis. However, micronodular cirrhosis may also result from other types
of liver injury (e.g., following jejunoileal bypass), and thus alcoholic
cirrhosis and micronodular cirrhosis are not necessarily synonymous.
Conversely, alcoholic cirrhosis may progress to macronodular cirrhosis with
time.
Etiology
Pathology and Pathogenesis
With
continued alcohol intake and destruction of hepatocytes, fibroblasts (including
activated hepatic stellate cells that have transformed into myofibroblasts with
contractile properties) appear at the site of injury and deposit collagen.
Weblike septa of connective tissue appear in periportal and pericentral zones
and eventually connect portal triads and central veins. This fine connective
tissue network surrounds small masses of remaining liver cells, which
regenerate and form nodules. Although regeneration occurs within the small
remnants of parenchyma, cell loss generally exceeds replacement. With
continuing hepatocyte destruction and collagen deposition, the liver shrinks in
size, acquires a nodular appearance, and becomes hard as "end-stage"
cirrhosis develops. Although alcoholic cirrhosis is usually a progressive
disease, appropriate therapy and strict avoidance of alcohol may arrest the
disease at most stages and permit functional improvement. In addition, there is
strong evidence that concomitant chronic hepatitis C virus (HCV) infection
significantly accelerates development of alcoholic cirrhosis.
Clinical Features
Signs and Symptoms
Alcoholic
cirrhosis may be clinically silent, and many cases (10 to 40%) are discovered
incidentally at laparotomy or autopsy. In many cases symptoms are insidious in
onset, occurring usually after 10 or more years of excessive alcohol use and progressing
slowly over subsequent weeks and months. Anorexia and malnutrition lead to
weight loss and a reduction in skeletal muscle mass. The patient may experience
easy bruising, increasing weakness, and fatigue. Eventually the clinical
manifestations of hepatocellular dysfunction and portal hypertension ensue,
including progressive jaundice, bleeding from gastroesophageal varices,
ascites, and encephalopathy. The abrupt onset of one of these complications may
be the first event prompting the patient to seek medical attention. In other
cases, cirrhosis first becomes evident when the patient requires treatment of
symptoms related to alcoholic hepatitis.
A
firm, nodular liver may be an early sign of disease; the liver may be either
enlarged, normal, or decreased in size. Other frequent findings include
jaundice, palmar erythema, spider angiomas, parotid and lacrimal gland
enlargement, clubbing of fingers, splenomegaly, muscle wasting, and ascites
with or without peripheral edema. Men may have decreased body hair and/or
gynecomastia and testicular atrophy, which, like the cutaneous findings, result
from disturbances in hormonal metabolism, including increased peripheral
formation of estrogen due to diminished hepatic clearance of the precursor
androstenedione. Testicular atrophy may reflect hormonal abnormalities or the
toxic effect of alcohol on the testes. In women, signs of virilization or
menstrual irregularities may occasionally be encountered. Dupuytren's
contractures resulting from fibrosis of the palmar fascia with resulting
flexion contracture of the digits are associated with alcoholism but are not
specifically related to cirrhosis.
Although
the cirrhotic patient may stabilize if drinking is discontinued, over a period
of years, the patient may become emaciated, weak, and chronically jaundiced.
Ascites and other signs of portal hypertension may become increasingly
prominent. Ultimately, most patients with advanced cirrhosis die in hepatic
coma, commonly precipitated by hemorrhage from esophageal varices or
intercurrent infection. Progressive renal dysfunction often complicates the
terminal phase of the illness.
Laboratory Findings
In
advanced alcoholic liver disease, abnormalities of laboratory tests are more
common. Anemia may result from acute and chronic gastrointestinal blood loss,
coexistent nutritional deficiency (notably of folic acid and vitamin B12),
hypersplenism, and a direct suppressive effect of alcohol on the bone marrow.
Hemolytic anemia, presumably due to effects of hypercholesterolemia or
erythrocyte membranes resulting in unusual spurlike projections
(acanthocytosis), has been described in some alcoholics with cirrhosis. Mild or
pronounced hyperbilirubinemia may be found, usually in association with varying
elevations of serum alkaline phosphatase levels. Levels of serum asparate
aminotransferase (AST) are frequently elevated, but levels >5![]() kat
(300 units) are unusual and should prompt one to look for other coincident or
complicating factors. In contrast to viral hepatitis, the serum AST is usually
disproportionately elevated relative to alanine aminotransferase (ALT), i.e.,
AST/ALT ratio >2. This discrepancy in alcoholic liver disease may result
from the proportionally greater inhibition of ALT synthesis by ethanol, which
may be partially reversed by pyridoxal phosphate.
kat
(300 units) are unusual and should prompt one to look for other coincident or
complicating factors. In contrast to viral hepatitis, the serum AST is usually
disproportionately elevated relative to alanine aminotransferase (ALT), i.e.,
AST/ALT ratio >2. This discrepancy in alcoholic liver disease may result
from the proportionally greater inhibition of ALT synthesis by ethanol, which
may be partially reversed by pyridoxal phosphate.
The
serum prothrombin time is frequently prolonged, reflecting reduced synthesis of
clotting proteins, most notably the vitamin K-dependent factors (see
"Coagulopathy," below). The serum albumin level is usually depressed,
while serum globulins are increased. Hypoalbuminemia reflects in part overall
impairment in hepatic protein synthesis, while hyperglobulinemia is thought to
result from nonspecific stimulation of the reticuloendothelial system. Elevated
blood ammonia levels in patients with hepatic encephalopathy reflect diminished
hepatic clearance because of impaired liver function and shunting of portal
venous blood around the cirrhotic liver into the systemic circulation (see
"Hepatic Encephalopathy," below).
A
variety of metabolic disturbances may be detected. Glucose intolerance due to
endogenous insulin resistance may be present; however, clinical diabetes is
uncommon. Central hyperventilation may lead to respiratory alkalosis in
patients with cirrhosis. Dietary deficiency and increased urinary losses lead
to hypomagnesemia and hypophosphatemia. In patients with ascites and dilutional
hyponatremia, hypokalemia may occur from increased urinary potassium losses due
in part to hyperaldosteronism. Prerenal azotemia is also observed in such
patients.
Diagnosis
Alcoholic
cirrhosis should be strongly suspected in patients with a history of prolonged
or excessive alcohol intake and physical signs of chronic liver disease.
However, since only 10 to 15% of individuals with excessive alcohol intake
develop cirrhosis, other causes and types of liver disease may have to be
excluded. The clinical features and laboratory findings are usually sufficient
to provide reasonable indication of the presence and extent of hepatic injury.
Although a percutaneous needle biopsy of the liver is not usually necessary to
confirm the typical findings of alcoholic hepatitis or cirrhosis, it may be
helpful in distinguishing patients with less advanced liver disease from those
with cirrhosis and in excluding other forms of liver injury such as viral
hepatitis. Biopsy may also be helpful as a diagnostic tool in evaluating
patients with clinical findings suggestive of alcoholic liver disease who deny
alcohol intake. In patients with features of cholestasis, ultrasonography may
be appropriate to exclude the presence of extrahepatic biliary obstruction.
When the clinical status of an otherwise stable cirrhotic patient deteriorates
without an obvious explanation, complicating conditions, such as infection,
portal vein thrombosis, and hepatocellular carcinoma, should be sought.
Prognosis
Abstinence
from alcohol as well as early and appropriate medical care can decrease
long-term morbidity and mortality and delay or prevent the appearance of
further complications. Patients who have had a major complication of cirrhosis
and who continue to drink have a 5-year survival of less than 50%. However,
those patients who remain abstinent have a substantially better prognosis. In
general, the overall outlook in patients with advanced liver disease remains
poor; most of these patients eventually die as a result of massive variceal
hemorrhage and/or profound hepatic encephalopathy.
![]() Treatment
Treatment
Alcoholic
cirrhosis is a serious illness that requires long-term medical supervision and
careful management. Therapy of the underlying liver disease is largely
supportive. Specific treatment is directed at particular complications such as
variceal bleeding and ascites (see below). While some studies suggest that
administration of glucocorticoids in moderately large doses for 4 weeks is
helpful in patients with severe alcoholic hepatitis and encephalopathy, these
drugs have no role in the treatment of established alcoholic cirrhosis. While
one study suggested a mortality benefit for the antifibrotic agent colchicine
in alcoholic cirrhosis, it has not yet been reproduced; thus colchicine cannot
be routinely recommended.
The
patient should be made to realize that there is no medication that will protect
the liver against the effects of further alcohol ingestion. Therefore, alcohol
should be absolutely forbidden. An important component of the complete care of
such patients is encouragement to become involved in an appropriate alcohol
counseling program.
All
medicines must be administered with caution in the patient with cirrhosis,
especially those eliminated or modified through hepatic metabolism or biliary
pathways. In particular, care must be taken to avoid overzealous use of drugs
that may directly or indirectly precipitate complications of cirrhosis. For
example, vigorous treatment of ascites with diuretics may result in electrolyte
abnormalities or hypovolemia, which can lead to coma. Similarly, even modest
doses of sedatives can lead to deepening encephalopathy. Aspirin should be
avoided in patients with cirrhosis because of its effects on coagulation and
gastric mucosa. Acetaminophen should be used with caution and in doses of less
than 2 g/day. Patients who drink alcohol are more sensitive to the hepatotoxic
effects of acetaminophen, probably due to increased metabolism of the drug to
toxic intermediates and decreased glutathione levels.
Posthepatitic and Cryptogenic Cirrhosis
Definition
Posthepatitic
or postnecrotic cirrhosis represents the final common pathway of many types of
chronic liver disease. Coarsely nodular and multilobular cirrhosis
are terms synonymous with posthepatitic cirrhosis. The term cryptogenic
cirrhosis has been used interchangeably with postpathepatitic cirrhosis,
but this designation should be reserved for those cases in which the etiology
of cirrhosis is unknown (approximately 10% of all patients with cirrhosis).
Etiology
Posthepatitic cirrhosis is a morphologic term referring to a
defined stage of advanced chronic liver injury of both specific and unknown
(cryptogenic) causes. Epidemiologic and serologic evidence suggest that viral
hepatitis (hepatitis B or hepatitis C) may be an antecedent factor in from
one-fourth to three-fourths of cases of apparently cryptogenic posthepatitic
cirrhosis. In areas where hepatitis B virus (HBV) infection is endemic (e.g.,
Southeast Asia, sub-Saharan Africa), up to 15% of the population may acquire
the infection in early childhood, and cirrhosis may ultimately develop in
one-fourth of these chronic carriers. Although HBV infection is much less
prevalent in the United States, it is relatively common among certain high-risk
groups (e.g., persons with multiple sexual partners, especially men who have
sex with men, injection drug users) and contributes to an increased incidence
of cirrhosis. In the United States, HCV infection accounts for many cases of
cirrhosis following blood transfusions. Before routine screening of blood
donors was introduced, hepatitis C occurred in 5 to 10% of blood recipients.
Following infection, cirrhosis may ultimately develop in more than 20% of
individuals after 20 years. More than half of patients who would previously
have been designated as having cryptogenic chronic liver disease have evidence
of HCV infection. Increasing recognition of the progressive nature of nonalcoholic
steatohepatitis has revealed that a large portion of cases previously
designated cryptogenic cirrhosis may be attributable to this disorder (Chap.
300). Posthepatitic cirrhosis may also develop in patients with autoimmune
hepatitis (Chap. 297).
The
most common causes of cirrhosis in the United States, which ultimately lead to
liver transplantation, include chronic HCV infection, alcohol, primary biliary
cirrhosis, primary sclerosing cholangitis, and nonalcoholic steatohepatitis
(NASH). Less common causes of posthepatitic cirrhosis, including drugs and
toxins, are listed in Table 299-1.
Table 299-1: Causes of Cirrhosis and/or Chronic
Liver Disease
|
Pathology
The
posthepatitic liver is typically shrunken in size, distorted in shape, and
composed of nodules of liver cells separated by dense and broad bands of
fibrosis. The microscopic picture is consistent with the gross impression.
Posthepatitic cirrhosis is characterized morphologically by (1) extensive
confluent loss of liver cells, (2) stromal collapse and fibrosis resulting in
broad bands of connective tissue containing the remains of many portal triads,
and (3) irregular nodules of regenerating hepatocytes, varying in size from
microscopic to several centimeters in diameter.
Clinical Features
In
patients with cirrhosis of known etiology in whom there is progression to a
posthepatitic stage, the clinical manifestations are an extension of those resulting
from the initial disease process. Usually clinical symptoms are related to
portal hypertension and its sequelae, such as ascites, splenomegaly,
hypersplenism, encephalopathy, and bleeding gastroesophageal varices. The
hematologic and liver function abnormalities resemble those seen with other
types of cirrhosis. In a few patients with posthepatitic cirrhosis, the
diagnosis may be made incidentally at operation, at postmortem, or by a needle
biopsy of the liver performed to investigate abnormal liver function tests or
hepatomegaly.
Diagnosis and Prognosis
Posthepatitic
cirrhosis should be suspected in patients with signs and symptoms of cirrhosis
or portal hypertension. Needle or operative liver biopsies confirm the
diagnosis, although nonuniformity of the pathologic process may result in
sampling errors. The diagnosis of cryptogenic cirrhosis is reserved for those
patients in whom no known etiology can be demonstrated. About 75% of patients
have progressive disease despite supportive therapy and die within 1 to 5 years
from complications, including variceal hemorrhage, hepatic encephalopathy, or
superimposed hepatocellular carcinoma.
![]() Treatment
Treatment
Management
is usually limited to treatment of the complications of portal hypertension,
including control of ascites, avoidance of drugs or excessive protein intake
that may induce hepatic coma, and prompt treatment of infections (see below).
In patients with asymptomatic cirrhosis, expectant management alone is
appropriate. In those patients in whom posthepatitic cirrhosis has developed as
a result of a treatable condition, therapy directed at the primary disorder may
limit further progression (e.g., Wilson's disease, hemochromatosis).
Biliary Cirrhosis
Biliary
cirrhosis results from injury to or prolonged obstruction of either the
intrahepatic or extrahepatic biliary system. It is associated with impaired
biliary excretion, destruction of hepatic parenchyma, and progressive fibrosis.
Primary biliary cirrhosis (PBC) is characterized by chronic inflammation and
fibrous obliteration of intrahepatic bile ductules. Secondary biliary cirrhosis
(SBC) is the result of long-standing obstruction of the larger extrahepatic
ducts. Although primary and secondary biliary cirrhosis are separate
pathophysiologic entities with respect to the initial insult, many clinical
features are similar.
Primary Biliary Cirrhosis
Etiology and Pathogenesis
The
cause of PBC remains unknown. Several observations suggest that a disordered
immune response may be involved. PBC is frequently associated with a variety of
disorders presumed to be autoimmune in nature, such as the syndrome of calcinosis,
Raynaud's phenomenon, esophageal dysmotility, sclerodactyly,
telangiectasia (CREST); the sicca syndrome (dry eyes and dry mouth);
autoimmune thyroiditis; type 1 diabetes mellitus; and IgA deficiency.
Most
important, a circulating IgG antimitochondrial antibody (AMA) is detected in
more than 90% of patients with PBC and only rarely in other forms of liver
disease. It has been demonstrated that these autoantibodies recognize three to
five inner mitochondrial membrane proteins identified as enzymes of the
pyruvate dehydrogenase complex (PDC), the branched chain-ketoacid dehydrogenase
complex (BCKDC), and the ![]() -ketoglutarate
dehydrogenase complex (KGDC). The major autoantigen in PBC (found in 90% of
patients) has been identified as the 74-kDa E2 component of the PDC,
dihydrolipoamide acetyltransferase. The antibodies are directed to a region
essential for binding of a lipoic acid cofactor and inhibit the overall enzymatic
activity of the PDC. Other AMA autoantibodies in PNC patients are directed to
similar constituents of BCKDC and KGDC and also inhibit their enzymatic
function. It remains unclear whether these properties have a direct
pathogenetic role in the development of PBC. In addition to AMA, elevated serum
levels of IgM and cryoproteins consisting of immune complexes capable of
activating the alternative complement pathway are found in 80 to 90% of
patients. Aberrant expression of major histocompatibility complex class II
molecules has been found on biliary epithelium in association with PBC,
suggesting that these cells may serve as antigen-presenting cells in this
setting. Lymphocytes are prominent in the portal regions and surround damaged
bile ducts. These histologic findings resemble those noted in graft-versus-host
disease following bone marrow transplantation and suggest that damage to bile
ducts may be immunologically mediated, perhaps reflecting a defect in a
suppressor cell population.
-ketoglutarate
dehydrogenase complex (KGDC). The major autoantigen in PBC (found in 90% of
patients) has been identified as the 74-kDa E2 component of the PDC,
dihydrolipoamide acetyltransferase. The antibodies are directed to a region
essential for binding of a lipoic acid cofactor and inhibit the overall enzymatic
activity of the PDC. Other AMA autoantibodies in PNC patients are directed to
similar constituents of BCKDC and KGDC and also inhibit their enzymatic
function. It remains unclear whether these properties have a direct
pathogenetic role in the development of PBC. In addition to AMA, elevated serum
levels of IgM and cryoproteins consisting of immune complexes capable of
activating the alternative complement pathway are found in 80 to 90% of
patients. Aberrant expression of major histocompatibility complex class II
molecules has been found on biliary epithelium in association with PBC,
suggesting that these cells may serve as antigen-presenting cells in this
setting. Lymphocytes are prominent in the portal regions and surround damaged
bile ducts. These histologic findings resemble those noted in graft-versus-host
disease following bone marrow transplantation and suggest that damage to bile
ducts may be immunologically mediated, perhaps reflecting a defect in a
suppressor cell population.
Pathology
PBC
is divided into four stages based on morphologic findings. The earliest
recognizable lesion (stage I), termed chronic nonsuppurative destructive
cholangitis, is a necrotizing inflammatory process of the portal triads. It
is characterized by destruction of medium and small bile ducts, a dense
infiltrate of acute and chronic inflammatory cells, mild fibrosis, and
occasionally, bile stasis. At times, periductal granulomas and lymph follicles
are found adjacent to affected bile ducts. Subsequently, the inflammatory
infiltrate becomes less prominent, the number of bile ducts is reduced, and
smaller bile ductules proliferate (stage II). Progression over a period of
months to years leads to a decrease in interlobular ducts, loss of liver cells,
and expansion of periportal fibrosis into a network of connective tissue scars
(stage III). Ultimately, cirrhosis, which may be micronodular or macronodular,
develops (stage IV).
Clinical Features
Signs and Symptoms
Many
patients with PBC are asymptomatic, and the disease is initially detected on
the basis of elevated serum alkaline phosphatase levels during routine
screening. The majority of such patients remain asymptomatic for prolonged
periods, although most ultimately develop progressive liver injury.
Among
patients with symptomatic disease, 90% are women age 35 to 60. Often the
earliest symptom is pruritus, which may be either generalized or limited
initially to the palms and soles. In addition, fatigue is commonly a prominent
early symptom. After several months or years, jaundice and gradual darkening of
the exposed areas of the skin (melanosis) may ensue. Other early clinical
manifestations of PBC reflect impaired bile excretion. These include
steatorrhea and the malabsorption of lipid-soluble vitamins. Protracted elevation
of serum lipids, especially cholesterol, leads to subcutaneous lipid deposition
around the eyes (xanthelasmas) and over joints and tendons (xanthomas). Over a
period of months to years, the itching, jaundice, and hyperpigmentation slowly
worsen. Eventually, signs of hepatocellular failure and portal hypertension
develop and ascites appears. Progression may be quite variable. Whereas a
proportion of asymptomatic patients may show no signs of progression for a
decade or longer, in others, death due to hepatic insufficiency may occur
within 5 to 10 years after the first signs of the illness. Such decompensation
is often precipitated by uncontrolled variceal hemorrhage or infection.
Physical
examination may be entirely normal in the early phase of the disease, when
patients are asymptomatic or pruritus is the sole complaint. Later, there may
be jaundice of varying intensity, hyperpigmentation of the exposed skin areas,
xanthelasmas and tendinous and planar xanthomas, moderate to striking
hepatomegaly, splenomegaly, and clubbing of the fingers. Bone tenderness, signs
of vertebral compression, ecchymoses, glossitis, and dermatitis may all be
noted. Clinical evidence of the sicca syndrome can be found in as many as 75%
of patients, and serologic evidence of autoimmune thyroid disease in 25%. Other
conditions encountered with increased frequency include rheumatoid arthritis,
CREST syndrome, keratoconjunctivitis sicca, IgA deficiency, type 1 diabetes
mellitus, scleroderma, pernicious anemia, and renal tubular acidosis. Bone
disease is often a significant problem encountered over the course of the
disease. While osteomalacia occurs due to diminished vitamin D absorption,
accelerated osteoporosis in this patient population (the majority of whom are
postmenopausal women) is even more common.
Laboratory Findings
PBC
is increasingly diagnosed at a presymptomatic stage, prompted by the finding of
a twofold or greater elevation of the serum alkaline phosphatase during routine
screening. Serum 5′-nucleotidase activity and ![]() -glutamyl
transpeptidase levels are also elevated. In this setting, serum bilirubin is
usually normal and aminotransferase levels minimally increased. The diagnosis
is supported by a positive AMA test (titer > 1:40). The latter is both
relatively specific and sensitive; a positive test is found in over 90% of
symptomatic patients and is present in fewer than 5% of patients with other
liver diseases. As the disease evolves, the serum bilirubin level rises
progressively and may reach 510
-glutamyl
transpeptidase levels are also elevated. In this setting, serum bilirubin is
usually normal and aminotransferase levels minimally increased. The diagnosis
is supported by a positive AMA test (titer > 1:40). The latter is both
relatively specific and sensitive; a positive test is found in over 90% of
symptomatic patients and is present in fewer than 5% of patients with other
liver diseases. As the disease evolves, the serum bilirubin level rises
progressively and may reach 510 ![]() mol/L
(30 mg/dL) or more in the final stages. Serum aminotransferase values rarely
exceed 2.5 to 3.3
mol/L
(30 mg/dL) or more in the final stages. Serum aminotransferase values rarely
exceed 2.5 to 3.3 ![]() kat
(150 to 200 units). Hyperlipidemia is common, and a striking increase of the
serum unesterified cholesterol is often noted. An abnormal serum lipoprotein
(lipoprotein X) may be present in PBC but is not specific and appears in other
cholestatic conditions. A deficiency of bile salts in the intestine leads to
moderate steatorrhea and impaired absorption of the fat-soluble vitamins and
hypoprothrombinemia. Patients with PBC have elevated liver copper levels, but
this finding is not specific and is found in all disorders in which there is
prolonged cholestasis.
kat
(150 to 200 units). Hyperlipidemia is common, and a striking increase of the
serum unesterified cholesterol is often noted. An abnormal serum lipoprotein
(lipoprotein X) may be present in PBC but is not specific and appears in other
cholestatic conditions. A deficiency of bile salts in the intestine leads to
moderate steatorrhea and impaired absorption of the fat-soluble vitamins and
hypoprothrombinemia. Patients with PBC have elevated liver copper levels, but
this finding is not specific and is found in all disorders in which there is
prolonged cholestasis.
Diagnosis
PBC
should be considered in middle-aged women with unexplained pruritus or an
elevated serum alkaline phosphatase and in whom there may be other clinical or
laboratory features of protracted impairment of biliary excretion. Although a
positive serum AMA determination provides important diagnostic evidence,
false-positive results do occur; therefore, liver biopsy should be performed to
confirm the diagnosis. Rarely, the AMA test may be negative in patients with
histologic features of PBC. Frequently, patients have antibodies to the E2
protein in tests using these specific antigens. In some cases with histologic
features of PBC and a negative AMA, antinuclear or smooth-muscle antibodies are
present (as in autoimmune hepatitis), and the designation autoimmune
cholangitis is applied. The natural history of this entity, however,
appears to resemble that of PBC. If the AMA test is negative, the biliary tract
should be evaluated to exclude primary sclerosing cholangitis and remediable
extrahepatic biliary tract obstruction, especially in view of the frequent
presence of coexisting cholelithiasis.
![]() Treatment
Treatment
While
there is no specific therapy for PBC, ursodiol has been shown to improve
biochemical and histologic features and might improve survival, particularly
liver transplantation-free survival (although this remains unproven). Ursodiol
should be given in doses of 10 to 15 mg/kg per day, but lower doses are
sometimes just as effective in reducing serum alkaline phosphatase and
aminotransferase levels. Ursodiol should be given with food and can be taken in
a single dose daily. Side effects are rare: gastrointestinal intolerance
(bloating, indigestion) and skin rashes occur but are uncommon. Isolated
instances of severe exacerbation of pruritus have been reported in patients
with advanced disease. Ursodiol probably works by replacing the endogenously
produced hydrophobic bile acids with urosdeoxycholate, a hydrophilic and
relatively nontoxic bile acid.
Unfortunately,
ursodiol does not prevent ultimate progression of PBC, and the only established
"cure" is liver transplantation. Results of liver transplantation for
PBC are excellent, survival exceeding that for patients receiving
transplantation for most other forms of end-stage liver disease. Recurrence of
PBC after liver transplantation has been reported but is uncommon, and the
recurrent disease is only slowly progressive. Most patients remain AMA positive
after transplantation, and as many as 25% will have histologic features of PBC
on liver biopsy after 5 years. Other therapies such as glucocorticoids,
colchicine, methotrexate, azathioprine, cyclosporine, and tacrolimus have been
reported as effective in small cases series, but none have shown to be
effective in adequately controlled trials.
Relief
of symptoms is also an important part of management of PBC. As noted, ursodiol
may be helpful in controlling symptoms and improving the patient's sense of
well-being. Although the mechanism of the protracted pruritus is not entirely
clear, cholestyramine, an oral bile salt-sequestering resin, may be helpful in
doses of 8 to 12 g/d to decrease both pruritus and hypercholesterolemia.
Rifampin, opiate antagonists, ondansetron, plasmapheresis, and ultraviolet
light have all been tried for control of pruritus, with varying results.
Steatorrhea can be reduced by a low-fat diet and substituting medium-chain
triglycerides for dietary long-chain triglycerides. Fat-soluble vitamins A and
K should be given by parenteral injection at regular intervals to prevent or
correct night blindness and hypoprothrombinemia, respectively. Zinc
supplementation may be necessary if night blindness is refractory to vitamin A
therapy. An important part of management of PBC and any cholestatic liver
disease is assessment and treatment of osteoporosis and osteomalacia. Patients
should be screened periodically by bone densitometry and treated as needed with
calcium supplements, estrogen, and/or the newer bisphosphonate agents (e.g.,
alendronate). Progression of PBC leads to the typical complications of advanced
liver disease (see below).
Secondary Biliary Cirrhosis
Etiology
SBC
results from prolonged partial or total obstruction of the common bile duct or
its major branches. In adults, obstruction is most frequently caused by
postoperative strictures or gallstones, usually with superimposed infectious
cholangitis. Chronic pancreatitis may lead to biliary stricture and secondary
cirrhosis. SBC is also an important complication of primary sclerosing
cholangitis, a progressive immunologic disorder of the intrahepatic and
extrahepatic biliary tree (Chap. 302). Patients with malignant tumors of the
common bile duct or pancreas rarely survive long enough to develop SBC. In
children, congenital biliary atresia and cystic fibrosis are common causes of
SBC. Choledochal cysts, if unrecognized, may also be a rare cause of SBC.
Pathology and Pathogenesis
Unrelieved
obstruction of the extrahepatic bile ducts leads to (1) bile stasis and focal
areas of centrilobular necrosis followed by periportal necrosis, (2)
proliferation and dilatation of the portal bile ducts and ductules, (3) sterile
or infected cholangitis with accumulation of polymorphonuclear infiltrates
around bile ducts, and (4) progressive expansion of portal tracts by edema and
fibrosis. Extravasation of bile from ruptured interlobular bile ducts into
areas of periportal necrosis leads to the formation of "bile lakes"
surrounded by cholesterol-rich pseudoxanthomatous cells. As in other forms of
cirrhosis, injury is accompanied by regeneration in residual parenchyma. These
changes gradually lead to a finely nodular cirrhosis. In general, at least 3 to
12 months is required for biliary obstruction to result in cirrhosis. Relief of
the obstruction is frequently accompanied by biochemical and morphologic
improvement.
Clinical Features
The
symptoms, signs, and biochemical findings of SBC are similar to those of PBC.
Jaundice and pruritus are usually the most prominent features. In addition,
fever and/or right upper quadrant pain, reflecting bouts of cholangitis or
biliary colic, are typical. The manifestations of portal hypertension are found
only in advanced cases. SBC should be considered in any patient with clinical
and laboratory evidence of prolonged obstruction to bile flow, especially when
there is a history of previous biliary tract surgery or gallstones, bouts of
ascending cholangitis, or right upper quadrant pain. Cholangiography (either
percutaneous or endoscopic) usually demonstrates the underlying pathologic
process. Liver biopsy, although not always necessary from a clinical
standpoint, can document the development of cirrhosis.
![]() Treatment
Treatment
Relief
of obstruction to bile flow, by either endoscopic or surgical means, is the
most important step in the prevention and therapy of SBC. Effective decompression
of the biliary tract results in a significant improvement in both symptoms and
survival, even in patients with established cirrhosis. When obstruction cannot
be relieved, as in sclerosing cholangitis, antibiotics may be helpful acutely
in controlling superimposed infection or, when administered on a chronic basis,
as prophylactic therapy in suppressing recurring episodes of ascending
cholangitis. Without relief of obstruction, there is a steady progression to
end-stage cirrhosis and its terminal manifestations
Cardiac Cirrhosis
Definition
Prolonged,
severe right-sided congestive heart failure may lead to chronic liver injury
and cardiac cirrhosis. The characteristic pathologic features of fibrosis and
regenerative nodules distinguish cardiac cirrhosis from both reversible passive
congestion of the liver due to acute heart failure and acute hepatocellular
necrosis ("ischemic hepatitis" or "shock liver") resulting
from systemic hypotension and hypoperfusion of the liver.
Etiology and Pathology
In
right-sided heart failure, retrograde transmission of elevated venous pressure
via the inferior vena cava and hepatic veins leads to congestion of the liver.
Hepatic sinusoids become dilated and engorged with blood, and the liver becomes
tensely swollen. With prolonged passive congestion and ischemia from poor
perfusion secondary to reduced cardiac output, necrosis of centrilobular
hepatocytes ensues and leads to fibrosis in these central areas. Ultimately,
centrilobular fibrosis develops, with collagen extending outward in a
characteristic stellate pattern from the central vein. Gross examination of the
liver shows alternating red (congested) and pale (fibrotic) areas, a pattern
often referred to as "nutmeg liver." Improvement in management of cardiac
disorders, particularly advances in surgical treatment, has reduced the
frequency of cardiac cirrhosis.
Clinical Features
A
range of abnormalities of liver function tests may be found, though none is
uniformly present. The serum bilirubin is usually only mildly increased and may
be predominantly either conjugated or unconjugated. Mild to moderate elevation
in alkaline phosphatase level and prothrombin time prolongation are sometimes
present. The AST level is typically mildly elevated but may be transiently very
high following a period of marked systemic hypotension (shock liver), when the
clinical picture can mimic acute viral or drug-induced hepatitis. In cases of
tricuspid insufficiency the liver may be pulsatile, but this finding disappears
as cirrhosis develops. With prolonged right-sided heart failure the liver
becomes enlarged, firm, and usually nontender. The signs and symptoms of heart
failure usually overshadow the liver disease. Bleeding from esophageal varices
is rare, but chronic encephalopathy may be prominent, with a waxing and waning
course reflecting variations in the severity of right-sided heart failure.
Ascites and peripheral edema, often primarily related to the underlying cardiac
dysfunction, may be worsened by the superimposed liver disease.
Diagnosis
The
presence of a firm, enlarged liver with signs of chronic liver disease in a
patient with valvular heart disease, constrictive pericarditis, or cor
pulmonale of long duration (>10 years) should suggest cardiac cirrhosis.
Liver biopsy can confirm the diagnosis but is usually contraindicated because
of coagulopathy or ascites. Coexistent chronic heart and liver disease should
also raise the possibility of hemochromatosis (Chap. 345), amyloidosis (Chap.
319), or other infiltrative diseases.
Budd-Chiari syndrome resulting from the occlusion of the hepatic
veins or inferior vena cava may be confused with acute congestive hepatomegaly.
In this condition the liver is grossly enlarged and tender, and severe
intractable ascites is present. However, signs and symptoms of heart failure
are notably absent. The most common cause is thrombosis of the hepatic veins,
often in the setting of polycythemia rubra vera, myeloproliferative syndromes,
paroxysmal nocturnal hemoglobinuria, oral contraceptive use, or other hypercoagulable
states; it may also result from invasion of the inferior vena cava by tumor,
such as renal cell or hepatocellular carcinoma. Idiopathic membranous
obstruction of the inferior vena cava is the most common cause of this syndrome
in Japan. Hepatic venography or liver biopsy showing centrilobular congestion
and sinusoidal dilatation in the absence of right-sided heart failure
establishes the diagnosis of Budd-Chiari syndrome. Venocclusive disease
affecting the sublobular branches of the hepatic veins and the hepatic venues
may result from hepatic irradiation, treatment with certain antineoplastic
agents, or ingestion of pyrrolidizine alkaloids present in some herbal teas
("bush tea disease") and can mimic congestive hepatomegaly.
![]() Treatment
Treatment
Prevention
or treatment of cardiac cirrhosis depends on the diagnosis and therapy of the
underlying cardiovascular disorder. Improvement in cardiac function frequently
results in improvement of liver function and stabilization of the liver
disease.
Metabolic, Hereditary, Drug-Related, and Other
Types of Cirrhosis (See
Table 299-1)
Cirrhosis
or hepatitis may result from a wide variety of other processes encompassing the
spectrum of etiologic factors listed in Table 299-2. Although some of these
disorders have distinctive clinical or morphologic features, the manifestations
of cirrhosis are largely independent of the underlying pathogenic mechanism.
Table 299-2: Some Causes of Noncirrhotic Hepatic
Fibrosis
|
Noncirrhotic Fibrosis of the Liver
Several
diseases, either congenital or acquired, may be associated with localized or
generalized hepatic fibrosis. They are distinguished from cirrhosis by the
absence of hepatocellular damage and the lack of nodular regenerative activity.
The clinical manifestations in such cases are largely secondary to portal
hypertension. The different types of these disorders are indicated in Table
299-2; with the exception of schistosomiasis in some regions of the world, all
these conditions are relatively rare.
Major Complications of Cirrhosis
The
clinical course of patients with advanced cirrhosis is often complicated by a
number of important sequelae that are independent of the etiology of the
underlying liver disease. These include portal hypertension and its
consequences (e.g., gastroesophageal varices and splenomegaly), ascites,
hepatic encephalopathy, spontaneous bacterial peritonitis, hepatorenal
syndrome, and hepatocellular carcinoma.
Portal Hypertension
Definition and Pathogenesis
Normal
pressure in the portal vein is low (5 to 10 mmHg) because vascular resistance
in the hepatic sinusoids is minimal. Portal hypertension (>10 mmHg) most
commonly results from increased resistance to portal blood flow. Because the
portal venous system lacks valves, resistance at any level between the right
side of the heart and splanchnic vessels results in retrograde transmission of
an elevated pressure. Increased resistance can occur at three levels relative
to the hepatic sinusoids: (1) presinusoidal, (2) sinusoidal, and (3)
postsinudoidal. Obstruction in the presinusoidal venous compartment may
be anatomically outside the liver (e.g., portal vein thrombosis) or within the
liver itself but at a functional level proximal to the hepatic sinusoids so
that the liver parenchyma is not exposed to the elevated venous pressure (e.g.,
schistosomiasis).
Postsinusoidal obstruction may also occur outside the liver at
the level of the hepatic veins (e.g., Budd-Chiari syndrome), the inferior vena
cava, or, less commonly, within the liver (e.g., venocclusive disease). When
cirrhosis is complicated by portal hypertension, the increased resistance is
usually sinusoidal. While distinctions between pre-, post-, and
sinusoidal processes are conceptually appealing, functional resistance to portal
flow in a given patient may occur at more than one level. Portal hypertension
may also arise from increased blood flow (e.g., massive splenomegaly or
arteriovenous fistulas), but the low outflow resistance of the normal liver
makes this a rare clinical problem.
Cirrhosis is the most common cause of portal hypertension
in the United States. Clinically significant portal hypertension is present in
>60% of patients with cirrhosis. Portal vein obstruction is the
second most common cause; it may be idiopathic or occur in association with
cirrhosis, infection, pancreatitis, or abdominal trauma. Portal vein thrombosis
may develop in a variety of hypercoagulable states including polycythemia vera;
essential thrombocythemia; deficiencies of protein C, protein S, or
antithrombin III; resistance to activated protein C (factor V Leiden); and a
mutation of the prothrombin gene (G20210A). Portal vein thrombosis may be
idiopathic, though some of these patients may have a subclinical
myeloproliferative disorder. Hepatic vein thrombosis (Budd-Chiari syndrome) and
hepatic venoocclusive disease are relatively infrequent causes of portal
hypertension (see above). Portal vein occlusion may result in massive
hematemesis from gastroesophageal varices, but ascites is usually found only
when cirrhosis is present. Noncirrhotic portal fibrosis (Table 299-2) accounts
for only a few cases of portal hypertension.
Clinical Features
The
major clinical manifestations of portal hypertension include hemorrhage from
gastroesophageal varices, splenomegaly with hypersplenism, ascites, and acute
and chronic hepatic encephalopathy. These are related, at least in part, to the
development of portal-systemic collateral channels. The absence of valves in
the portal venous system facilitates retrograde (hepatofugal) blood flow from
the high-pressure portal venous system to the lower-pressure systemic venous
circulation. Major sites of collateral flow involve the veins around
cardioesophageal junction (esophagogastric varices), the rectum (hemorrhoids),
retroperitoneal space, and the falciform ligament of the liver (periumbilical
or abdominal wall collaterals). Abdominal wall collaterals appear as tortuous
epigastric vessels that radiate from the umbilicus toward the xiphoid and rib
margins (caput medusae).
A
frequent marker of the presence of cirrhosis in a patient being followed for
chronic liver disease is a progressive decrease in platelet count. A low-normal
platelet count can be the first clue to progression to cirrhosis. Ultimately, a
marked decrease in platelets (to 30,000 to 60,000/![]() L)
and white blood cells can occur.
L)
and white blood cells can occur.
Diagnosis
In
patients with known liver disease, the development of portal hypertension is
usually revealed by the appearance of splenomegaly, ascites, encephalopathy,
and/or esophageal varices. Conversely, the finding of any of these features
should prompt evaluation of the patient for the presence of underlying portal
hypertension and liver disease. Varices are most reliably documented by
fiberoptic esophagoscopy; their presence lends indirect support to the
diagnosis of portal hypertension. Although rarely necessary, portal venous
pressure may be measured directly by percutaneous transhepatic "skinny
needle" catheterization or indirectly through transjugular cannulation of
the hepatic veins. Both free and wedged hepatic vein pressure should be
measured. While the latter is elevated in sinusoidal and postsinusoidal portal
hypertension, including cirrhosis, this measurement is usually normal in
presinusoidal portal hypertension. In patients in whom additional information
is necessary (e.g., preoperative evaluation before portal-systemic shunt surgery)
or when percutaneous catheterization is not feasible, mesenteric and hepatic
angiography may be helpful. Particular attention should be directed to the
venous phase to assess the patency of the portal vein and the direction of
portal blood flow.
![]() Treatment
Treatment
Although
treatment is usually directed toward a specific complication of portal
hypertension, attempts are sometimes made to reduce the pressure in the portal
venous system. Surgical decompression procedures have been used for many years
to lower portal pressure in patients with bleeding esophageal varices (see
below). However, portal-systemic shunt surgery does not result in improved survival
rates in patients with cirrhosis. Decompression can now be accomplished without
surgery through the percutaneous placement of a portal-systemic shunt, termed a
transjugular intrahepatic portosystemic shunt (TIPS). ![]() -Adrenergic
blockade with propranolol or nadolol reduces portal pressure through inhibition
of vasodilatory effects on both the splanchnic arterial bed and the portal
venous system in combination with reduced cardiac output. Such therapy has been
shown to be effective in preventing both a first variceal bleed and subsequent
episodes after an initial bleed. Treatment of patients with clinically
significant sequelae of portal hypertension, especially variceal bleeding, with
doses of propranolol titrated to reduce the resting pulse by 25% is reasonable
if no contraindications exist.
-Adrenergic
blockade with propranolol or nadolol reduces portal pressure through inhibition
of vasodilatory effects on both the splanchnic arterial bed and the portal
venous system in combination with reduced cardiac output. Such therapy has been
shown to be effective in preventing both a first variceal bleed and subsequent
episodes after an initial bleed. Treatment of patients with clinically
significant sequelae of portal hypertension, especially variceal bleeding, with
doses of propranolol titrated to reduce the resting pulse by 25% is reasonable
if no contraindications exist.
Vigorous
treatment of patients with alcoholic hepatitis and cirrhosis, chronic active
hepatitis, or other liver diseases may lead to a fall in portal pressure and to
a reduction in variceal size. In general, however, portal hypertension due to
cirrhosis is not reversible. In appropriately selected patients, hepatic
transplantation will be beneficial.
Variceal Bleeding
Pathogenesis
While
vigorous hemorrhage may arise from any portal-systemic venous collaterals,
bleeding is most common from varices in the region of the gastroesophageal
junction. The factors contributing to bleeding from gastroesophageal varices
are not entirely understood but include the degree of portal hypertension
(>12 mmHg) and the size of the varices.
Clinical Features and Diagnosis
Variceal
bleeding often occurs without obvious precipitating factors and usually presents
with painless but massive hematemesis with or without melena. Associated signs
range from mild postural tachycardia to profound shock, depending on the extent
of blood loss and degree of hypovolemia. Because patients with varices may
bleed just as frequently from other gastrointestinal lesions (e.g., peptic
ulcer, gastritis), exclusion of other bleeding sources is important even in
patients with prior variceal hemorrhage. Endoscopy is the best approach to
evaluate upper gastrointestinal hemorrhage in patients with known or suspected
portal hypertension.
![]() Treatment
Treatment
(See
Fig. 299-1) Variceal bleeding is a life-threatening emergency. Prompt estimation
and vigorous replacement of blood loss to maintain intravascular volume are
essential and take precedence over diagnostic studies and more specific
intervention to stop the bleeding. However, excessive fluid administration can
increase portal pressure with resultant further bleeding and should therefore
be avoided. Replacement of clotting factors with fresh-frozen plasma is
important in patients with coagulopathy. Patients are best managed in an
intensive care unit and require close monitoring of central venous or pulmonary
capillary wedge pressures, urine output, and mental status. Only when the
patient is hemodynamically stable should attention be directed toward specific
diagnostic studies (especially endoscopy) and other therapeutic modalities to
prevent further or recurrent bleeding.
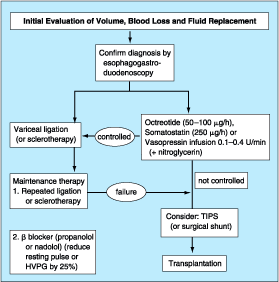
Figure 299-1: Approach to the patient with bleeding
esophageal varices. Use of a beta blocker is the only intervention demonstrated
to offer prophylactic benefit in a patient who has never bled. TIPS,
transjugular intrahepatic portosystemic shunt.
About
half of all episodes of variceal hemorrhage cease without intervention,
although the risk of rebleeding is very high. The medical management of acute
variceal hemorrhage includes the use of vasoconstrictors
(somatostatin/octreotide or vasopressin), balloon tamponade, and endoscopic
banding of varices or endoscopic sclerosis of varices (sclerotherapy).
Intravenous infusion of vasopressin at a rate of 0.1 to 0.4 U/min
results in generalized vasoconstriction leading to diminished blood flow in the
portal venous system. Intravenous infusion of vasopressin is as effective as
selective intraarterial administration. Control of bleeding can be achieved in
up to 80% of cases, but bleeding recurs in more than half after the vasopressin
is tapered and discontinued. Furthermore, a number of serious side effects,
including cardiac and gastrointestinal tract ischemia, acute renal failure, and
hyponatremia, may be associated with vasopressin therapy. Concurrent use of
venodilators such as nitroglycerin as an intravenous infusion or isosorbide
dinitrate sublingually may enhance the effectiveness of vasopressin and reduce
complications. Somatostatin and its analogue, octreotide, are
direct splanchnic vasoconstrictors. In some studies somatostatin, given as an
initial 250-![]() g
bolus followed by constant infusion (250
g
bolus followed by constant infusion (250 ![]() g/h),
has been found to be as effective as vasopressin. Octreotide at doses of 50 to
100
g/h),
has been found to be as effective as vasopressin. Octreotide at doses of 50 to
100 ![]() g/h
is also effective. These agents are preferable to vasopressin, offering
equivalent efficacy with fewer complications. If bleeding is too vigorous or
endoscopy is not available, balloon tamponade of the bleeding varices
may be accomplished with a triple-lumen (Sengstaken-Blakemore) or four-lumen
(Minnesota) tube with esophageal and gastric balloons. Because of the high risk
of aspiration, endotracheal intubation should be performed prior to placing one
of these tubes. After the tube is introduced into the stomach, the gastric
balloon is inflated and pulled back into the cardia of the stomach. If bleeding
does not stop, the esophageal balloon is inflated for additional tamponade.
Complications occur in 15% or more of patients and include aspiration
pneumonitis as well as esophageal rupture.
g/h
is also effective. These agents are preferable to vasopressin, offering
equivalent efficacy with fewer complications. If bleeding is too vigorous or
endoscopy is not available, balloon tamponade of the bleeding varices
may be accomplished with a triple-lumen (Sengstaken-Blakemore) or four-lumen
(Minnesota) tube with esophageal and gastric balloons. Because of the high risk
of aspiration, endotracheal intubation should be performed prior to placing one
of these tubes. After the tube is introduced into the stomach, the gastric
balloon is inflated and pulled back into the cardia of the stomach. If bleeding
does not stop, the esophageal balloon is inflated for additional tamponade.
Complications occur in 15% or more of patients and include aspiration
pneumonitis as well as esophageal rupture.
Where
available, endoscopic intervention should be employed as the first line
of treatment to control bleeding acutely (Chaps. 44 and 283). Over the past 18
years, endoscopic sclerosis of esophageal varices has been extensively
employed. In this procedure, the varices are injected with one of several
sclerosing agents via a needle-tipped catheter passed through the endoscope.
After endoscopic identification of varices as the presumed source of bleeding,
sclerotherapy controls acute bleeding in up to 90% of cases. In addition, repeated
sclerotherapy can be performed until obliteration of all varices is
accomplished in an effort to prevent recurrent bleeding. While available data
support the efficacy of sclerotherapy in controlling bleeding acutely and in
decreasing rebleeding rates, repeated sclerotherapy has not been documented to
prolong survival. Mucosal ulceration resulting from injection of the caustic
sclerosant may occur and result in further hemorrhage or stenosis. More
recently, endoscopic band ligation, in which esophageal varices are ligated and
strangulated with endoscopically placed small elastic O-rings, has gained
favor. Band ligation has proven to be at least as effective as sclerotherapy in
controlling acute variceal bleeding and preventing rebleeding. Because it has been
associated with fewer treatment-related complications, band ligation is
recommended for long-term obliteration of varices that have bled. Although
prophylactic sclerosis or banding of esophageal varices in the absence of
proven bleeding cannot yet be recommended, one report suggests that banding may
be more effective than beta-blockade in primary prevention of variceal bleeding
in high-risk patients.
The
effectiveness of nonselective ![]() -adrenergic
blocking agents (e.g., propranolol) in the management of acute variceal
bleeding is limited due to concomitant hypotension resulting from hypovolemia.
However, a number of studies suggest they may be of value in secondary
prevention of recurrent variceal hemorrhage. Moreover, prophylactic treatment
with nonselective beta blockers (propranolol or nadolol) in patients with large
("high-risk") varices that have never bled appears to decrease the
incidence of bleeding and prolong survival. Thus, endoscopic screening for
varices in patients with cirrhosis is desirable; some have suggested this
should be repeated every other year. Patients with portal hypertension without
specific contraindications should be given propranolol in doses that produce a
25% reduction in the resting heart rate or the hepatic venous pressure gradient
(HVPG), where available. Propranolol may also prevent recurrent bleeding from
severe portal hypertensive gastropathy in patients with cirrhosis. The optimal
combination of endoscopic and pharmacologic therapy for prevention of recurrent
hemorrhage remains to be established and is the subject of ongoing trials.
-adrenergic
blocking agents (e.g., propranolol) in the management of acute variceal
bleeding is limited due to concomitant hypotension resulting from hypovolemia.
However, a number of studies suggest they may be of value in secondary
prevention of recurrent variceal hemorrhage. Moreover, prophylactic treatment
with nonselective beta blockers (propranolol or nadolol) in patients with large
("high-risk") varices that have never bled appears to decrease the
incidence of bleeding and prolong survival. Thus, endoscopic screening for
varices in patients with cirrhosis is desirable; some have suggested this
should be repeated every other year. Patients with portal hypertension without
specific contraindications should be given propranolol in doses that produce a
25% reduction in the resting heart rate or the hepatic venous pressure gradient
(HVPG), where available. Propranolol may also prevent recurrent bleeding from
severe portal hypertensive gastropathy in patients with cirrhosis. The optimal
combination of endoscopic and pharmacologic therapy for prevention of recurrent
hemorrhage remains to be established and is the subject of ongoing trials.
Surgical
treatment of portal hypertension and variceal bleeding involves the creation of
a portal-systemic shunt to permit decompression of the portal system. Two types
of portal systemic shunts have been used: nonselective shunts, to
decompress the entire portal system, and selective shunts, intended to decompress
only the varices while maintaining blood flow to the liver itself. Nonselective
shunts include end-to-side or side-to-side portacaval and proximal splenorenal
anastomoses; selective shunts include the distal splenorenal shunt.
Nonselective shunts are more likely to be complicated by encephalopathy than
selective shunts. Emergency portal-systemic nonselective shunts may control
acute hemorrhage, but such surgery is usually used only as a last resort
because early operative mortality can be high. The role of portal-systemic
shunt surgery after initial control of bleeding by nonoperative means is also
uncertain. Surgically created shunts effectively reduce the risk of recurrent
hemorrhage, but the overall mortality of patients undergoing such surgery is
comparable to that of unoperated patients. Although patients who have undergone
portal-systemic surgery succumb to recurrent bleeding less commonly than
unoperated patients, this improvement is counterbalanced by increased morbidity
from encephalopathy and death from progressive liver failure. Increasingly,
therapeutic portal-systemic shunts have been reserved for patients who
experience further bleeding despite serial endoscopic sclerotherapy or band
ligation.
In
TIPS, a technique developed to create a portal-systemic shunt by a percutaneous
approach, an expandable metal stent is advanced to the hepatic veins under
angiographic guidance and then through the substance of the liver to create a
direct portacaval channel. This technique offers an alternative to surgery for
refractory bleeding due to portal hypertension. However, stents frequently
undergo stenosis or occlude over a period of months, prompting the need for a
second TIPS or an alternative approach. Encephalopathy may be encountered after
TIPS just as in the surgical shunts and is especially problematic in the
elderly and those patients with preexisting encephalopathy. TIPS should be
reserved for those individuals who fail endoscopic or medical management and
are poor surgical risks. TIPS may have a useful role as a "bridge"
for those patients with end-stage cirrhosis awaiting liver transplantation.
Procedures such as esophageal transection have also been advocated for the
management of acute variceal bleeding, but their efficacy remains unproven.
Even though recent trials found that esophageal transection was as effective as
endoscopic sclerotherapy, transection is usually considered a last resort.
The
management of bleeding gastric fundal varices, either alone or in conjunction
with esophageal varices, is more problematic, since sclerotherapy and banding
are generally not effective. Vasoactive pharmacologic therapy should be
instituted, but TIPS or shunt surgery should be considered because of high
failure and rebleeding rates. For isolated gastric varices, splenic vein
thrombosis should be specifically sought, since splenectomy is curative.
Portal Hypertensive Gastropathy
Although variceal hemorrhage is the most commonly
encountered bleeding complication of portal hypertension, many patients will develop
a congestive gastropathy due to the venous hypertension. In this condition,
identified by endoscopic examination, the mucosa appears engorged and friable.
Indolent mucosal bleeding occurs rather than the brisk hemorrhage typical of a
variceal source. ![]() -Adrenergic
blockade with propranolol (reducing splanchnic arterial pressure as well as
portal pressure) is sometimes effective in ameliorating this condition. H2
receptor antagonists or other agents useful in the treatment of peptic disease
are usually not helpful.
-Adrenergic
blockade with propranolol (reducing splanchnic arterial pressure as well as
portal pressure) is sometimes effective in ameliorating this condition. H2
receptor antagonists or other agents useful in the treatment of peptic disease
are usually not helpful.
Splenomegaly
Definition and Pathogenesis
Congestive
splenomegaly is common in patients with severe portal hypertension. Rarely,
massive splenomegaly from nonhepatic disease leads to portal hypertension due
to increased blood flow in the splenic vein.
Clinical Features
Although
usually asymptomatic, splenomegaly may be massive and contribute to the
thrombocytopenia or pancytopenia of cirrhosis. In the absence of cirrhosis,
splenomegaly in association with variceal hemorrhage should suggest the
possibility of splenic vein thrombosis.
![]() Treatment
Treatment
Splenomegaly
usually requires no specific treatment, although massive enlargement of the
spleen may occasionally necessitate splenectomy at the time of shunt surgery.
However, it should be noted that splenectomy without an accompanying shunt may
actually increase portal pressure, and portal vein thrombosis may result from
splenectomy. Splenectomy may also be indicated if splenomegaly is the cause
rather than the result of portal hypertension (as in splenic vein thrombosis).
Thrombcytopenia alone is rarely severe enough to necessitate removal of the
spleen. Splenectomy should be avoided in a patient eligible for liver
transplantation.
Ascites
Definition
Ascites
is the accumulation of excess fluid within the peritoneal cavity. It is most
frequently encountered in patients with cirrhosis and other forms of severe
liver disease, but a number of other disorders may lead to either transudative
or exudative ascites (Chap. 46).
Pathogenesis
The
accumulation of ascitic fluid represents a state of total-body sodium and water
excess, but the event that initiates this imbalance is unclear. Three theories
have been proposed (Fig. 299-2). The "underfilling" theory suggests
that the primary abnormality is inappropriate sequestration of fluid within the
splanchnic vascular bed due to portal hypertension and a consequent decrease in
effective circulating blood volume. According to this theory, an apparent
decrease in intravascular volume (underfilling) is sensed by the kidney, which
responds by retaining salt and water. The "overflow" theory suggests
that the primary abnormality is inappropriate renal retention of salt and water
in the absence of volume depletion. A third and more recent theory, the
peripheral arterial vasodilation hypothesis, may unify the earlier theories and
accounts for the constellation of arterial hypotension and increased cardiac
output in association with high levels of vasoconstrictor substances that are
routinely found in patients with cirrhosis and ascites. Again, sodium retention
is considered secondary to arterial vascular underfilling and the result of a
disproportionate increase of the vascular compartment due to arteriolar
vasodilation rather than from decreased intravascular volume. According to this
theory, portal hypertension results in splanchnic arteriolar vasodilation,
mediated by nitric oxide, and leading to underfilling of the arterial vascular
space and baroreceptor-mediated stimulation of renin-angiotensin, sympathetic
output, and antidiuretic hormone release.
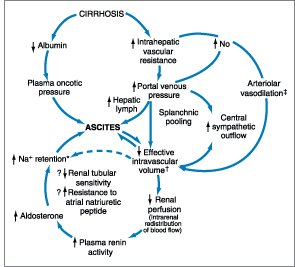
Figure 299-2: Multiple factors involved in development
of ascites. Current concepts suggest that initiating factor may be primary
sodium retention (*"overflow"), diminished effective intravascular
volume (![]() "underfilling"),
or arteriolar vasodilation (
"underfilling"),
or arteriolar vasodilation (![]() "vasodilation").
"vasodilation").
Regardless
of the initiating event, a number of factors contribute to accumulation of
fluid in the abdominal cavity (Fig. 299-2). Elevated levels of serum
epinephrine and norepinephrine have been well documented. Increased central
sympathetic outflow is found in patients with cirrhosis and ascites but not
in those with cirrhosis alone. Increased sympathetic output results in
diminished natriuresis by activation of the renin-angiotensin system and
diminished sensitivity to atrial natriuretic peptide. Portal hypertension
plays an important role in the formation of ascites by raising hydrostatic
pressure within the splanchnic capillary bed. Hypoalbuminemia and reduced
plasma oncotic pressure also favor the extravasation of fluid from plasma
to the peritoneal cavity, and thus ascites is infrequent in patients with
cirrhosis unless both portal hypertension and hypoalbuminemia are present.
Hepatic lymph may weep freely from the surface of the cirrhotic liver due to
distortion and obstruction of hepatic sinusoids and lymphatics and contributes
to ascites formation. In contrast to the contribution of transudative fluid
from the portal vascular bed, hepatic lymph may weep into the peritoneal cavity
even in the absence of marked hypoproteinemia because the endothelial lining of
the hepatic sinusoids is discontinuous. This mechanism may account for the high
protein concentration present in the ascitic fluid of some patients with
venoocclusive disease or the Budd-Chiari syndrome.
Renal factors also play an important role in perpetuating
ascites. Patients with ascites fail to excrete a water load in a normal
fashion. They have increased renal sodium reabsorption by both proximal and
distal tubules, the latter due largely to increased plasma renin activity and
secondary hyperaldosteronism. Insensitivity to circulating atrial natriuretic
peptide, often present in elevated concentrations in patients with cirrhosis
and ascites, may be an important contributory factor in many patients. This
insensitivity has been documented in those patients with the most severely
impaired sodium excretion, who typically also exhibit low arterial pressure and
marked overactivity of the renin-aldosterone axis. Renal vasoconstriction,
perhaps resulting from increased serum prostaglandin or catecholamine levels,
may also contribute to sodium retention. Recently a role for endothelin, a
potent vasoconstrictor peptide, has been proposed. While elevated levels have
been reported by some, this has not been observed by others.
As
discussed in Chap. 46, ascites may arise in a number of clinical settings in
addition to cirrhosis and portal hypertension. Although historically ascites
was classified as either transudative or exudative, similar to the
characterization of pleural fluids, this schema has limitations. Instead, the
serum-ascites albumin gradient (SAAG) provides a better classification than
total protein content or other parameters. In cirrhosis, the serum albumin
concentration is usually at least 10 g/L (1 g/dL) higher than that of the
ascitic fluid, thus yielding a high SAAG [![]() 11
g/L (
11
g/L (![]() 1.1
g/dL)], reflecting indirectly the abnormally high hydrostatic pressure gradient
between the portal bed and the ascitic compartment. Conversely, the presence of
a low SAAG [<11 g/L (<1.1 g/dL)] will usually exclude cirrhosis and
portal hypertension.
1.1
g/dL)], reflecting indirectly the abnormally high hydrostatic pressure gradient
between the portal bed and the ascitic compartment. Conversely, the presence of
a low SAAG [<11 g/L (<1.1 g/dL)] will usually exclude cirrhosis and
portal hypertension.
Clinical Features and Diagnosis
Usually
ascites is first noticed by the patient because of increasing abdominal girth.
More pronounced accumulation of fluid may cause shortness of breath because of
elevation of the diaphragm. When peritoneal fluid accumulation exceeds 500 mL,
ascites may be demonstrated on physical examination by the presence of shifting
dullness, a fluid wave, or bulging flanks. Ultrasound examination, preferably
with a Doppler study, can detect smaller quantities of ascites and should be
performed when physical examination is equivocal or when the cause of the
recent onset of ascites is not clear (e.g., exclude Budd-Chiari syndrome or
portal vein thrombosis).
![]() Treatment
Treatment
(See
Fig. 299-3) A thorough search should be made for precipitating factors in the
patient with recent onset of or worsening ascites, e.g., excessive salt intake,
medication noncompliance, superimposed infection, worsening liver disease,
portal vein thrombosis, or development of hepatocellular carcinoma. When
ascites develops in the setting of severe, acute liver disease, resolution of
ascites is likely to follow improvement in liver function. More commonly,
ascites develops in patients with stable or steadily worsening liver function.
Paracentesis should usually be performed with a small-gauge needle at the time
of initial evaluation or at the time of any clinical deterioration of a
cirrhotic patient with ascites. A small amount of fluid (<200 mL) should be
obtained and examined for evidence of infection, tumor, or other possible
causes and complications of ascites. Therapeutic intervention is indicated both
to prevent potential complications and to control progressive increase in
ascites, which may become pronounced enough to cause physical discomfort. For
the patient with a modest accumulation, therapy can be undertaken as an
outpatient and should be gentle and incremental (see below). The goal is the
loss of no more than 1.0 kg/d if both ascites and peripheral edema are present
and no more than 0.5 kg/d in patients with ascites alone. In some patients,
particularly those with a large accumulation of fluid, it may be desirable to
hospitalize the patient so that daily weights and frequent serum electrolyte
levels can be monitored and compliance ensured. Although abdominal girth
measurements are frequently used as an index of fluid loss, they tend to be
unreliable.
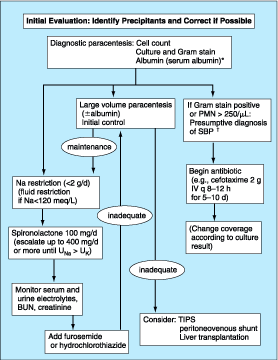
Figure 299-3: Approach to the patient with ascites [and
spontaneous bacterial peritonitis (SBP)]. PMN, polymorphonuclear leukocytes;
TIPS, transjugular intrahepatic portosystemic shunt. *Calculate SAAG [serum
ascites albumin gradient = serum albumin - ascites albumin (g/dL)] to confirm
high gradient (>1.1 g/dL) in portal hypertension. ![]() If
PMN >250/
If
PMN >250/![]() L
but culture is negative = culture negative neutrocytic ascites; begin empirical
antibiotic and retap at 48 h; if culture is positive but PMN < 250/
L
but culture is negative = culture negative neutrocytic ascites; begin empirical
antibiotic and retap at 48 h; if culture is positive but PMN < 250/![]() L
normal = monomicrobial nonneutrocytic bacterascites; retap; treat if PMN >
250/
L
normal = monomicrobial nonneutrocytic bacterascites; retap; treat if PMN >
250/![]() L:
if polymicrobial infection, exclude secondary peritonitis. UNa,
urinary sodium; UKa, urinary potassium; BUN, blood urea nitrogen.
L:
if polymicrobial infection, exclude secondary peritonitis. UNa,
urinary sodium; UKa, urinary potassium; BUN, blood urea nitrogen.
Salt
restriction is the cornerstone of therapy. A diet containing 800 mg sodium (2 g
NaCl) is often adequate to induce a negative sodium balance and permit
diuresis. Response to salt restriction alone is more likely to occur if the
ascites is of recent onset, the underlying liver disease is reversible, a
precipitating factor can be corrected, or the patient has a high urinary sodium
excretion (>25 mmol/d) and normal renal function. Fluid restriction of approximately
1000 mL/d does little to enhance diuresis but may be necessary to correct
hyponatremia. If sodium restriction alone fails to result in diuresis and
weight loss, diuretics should be prescribed. Because of the role of
hyperaldosteronism in sustaining salt retention, spironolactone or other distal
tubule-acting diuretics (triamterene, amiloride) are the drugs of choice. These
agents are also preferred because of their gentle action and specific
potassium-sparing properties. Spironolactone is initially given in a dose of
100 mg a day and is increased as needed by 100 mg/d every several days to a
maximum dose that should rarely exceed 400 mg/d. An indication of the minimum
effective dose of spironolactone may be obtained by monitoring urinary
electrolyte concentrations for a rise in sodium and fall in potassium levels,
reflecting effective competitive inhibition of aldosterone. Conversely, the
development of azotemia or hyperkalemia may be dose-limiting or even warrant a
reduction in the amount of this medication. In some patients, diuresis cannot
be initiated despite maximal doses of distal tubule-acting agents (e.g., 400 mg
spironolactone) because of avid proximal tubular sodium absorption. More potent
and proximally acting diuretics (furosemide, thiazide, or ethacrynic acid) may
then be added cautiously to the regimen. Spironolactone plus furosemide, 40 or
80 mg/d, is usually sufficient to initiate a diuresis in most patients.
However, such aggressive therapy must be used with great caution to avoid
plasma volume depletion, azotemia, and hypokalemia, which may lead to
encephalopathy.
In
patients with pronounced ascites, particularly those requiring hospitalization,
large-volume paracentesis has proven to be an effective and less costly
approach to initial management than prolonged bed rest and conventional
diuretic treatment. In this approach, ascitic fluid is removed by peritoneal
cannula using strict aseptic techniques and monitoring hemodynamic and renal
function. This can be safely accomplished in a single session. The need for
concomitant albumin replacement by intravenous infusion remains controversial
but may be prudent in the patient without peripheral edema, to avoid depleting
the intravascular space and precipitating hypotension. Maintenance diuretic therapy
in conjunction with sodium restriction may then be instituted to avoid
recurrent ascites.
A
minority of patients with advanced cirrhosis has "refractory ascites"
or rapidly reaccumulate fluid after control by paracentesis. In some patients,
a side-to-side portacaval shunt may result in improvement in ascites,
although generally these patients are extremely poor surgical risks. In the
past, intractable ascites has also been treated with the surgical implantation
of a plastic peritoneovenous shunt, which has a pressure-sensitive,
one-way valve allowing ascitic fluid to flow from the abdominal cavity to the
superior vena cava. However, the usefulness of this technique is limited by a
high rate of complications such as infection, disseminated intravascular
coagulation, and thrombosis of the shunt. More recently, in selected patients
TIPS has been used effectively to control refractory ascites, although portal
decompression, while mobilizing ascitic fluid, has precipitated severe hepatic
encephalopathy in some patients. TIPS remains a promising but unproven
treatment for refractory ascites. None of these shunts has been shown to extend
life expectancy.
Spontaneous Bacterial Peritonitis (SBP)
Patients
with ascites and cirrhosis may develop acute bacterial peritonitis without an
obvious primary source of infection. Patients with very advanced liver disease
are particularly susceptible to SBP. The ascitic fluid in these patients
typically has especially low concentrations of albumin and other so-called
opsonic proteins, which normally may provide some protection against bacteria.
Although key steps in the pathogenesis of SBP remain to be elucidated, it is
clear that most bacteria contributing to SBP derive from the bowel and
eventually are spread to ascitic fluid by the hematogenous route after
transmigration through the bowel wall and transversing the lymphatics. Clinical
features can include abrupt onset of fever, chills, generalized abdominal pain,
and, rarely, rebound abdominal tenderness. However, the clinical symptoms may
be minimal, and some patients manifest only worsening jaundice or
encephalopathy in the absence of localizing abdominal complaints. The diagnosis
is based on careful examination of the ascitic fluid. An ascitic fluid
leukocyte count of >500 cells/L (with a proportion of polymorphonuclear
leukocytes of ![]() 50%)
or more than 250 polymorphonuclear leukocytes should suggest the possibility of
bacterial peritonitis while results of bacterial cultures of ascitic fluid are
pending. Other measurements such as fluid pH or determination of gradients
between serum and fluid pH or lactate are generally not necessary. The presence
of more than 10,000 leukocytes per liter, multiple organisms, or failure to
improve after standard therapy for 48 h suggest that the peritonitis may be
secondary to an infection elsewhere in the body.
50%)
or more than 250 polymorphonuclear leukocytes should suggest the possibility of
bacterial peritonitis while results of bacterial cultures of ascitic fluid are
pending. Other measurements such as fluid pH or determination of gradients
between serum and fluid pH or lactate are generally not necessary. The presence
of more than 10,000 leukocytes per liter, multiple organisms, or failure to
improve after standard therapy for 48 h suggest that the peritonitis may be
secondary to an infection elsewhere in the body.
A
variant of SBP, designated monomicrobial nonneutrocytic bacterascites,
is sometimes seen. In these patients, culture of ascitic fluid yields bacteria,
but the neutrophil count is less than 250/L. These patients often have less
severe liver disease than those found initially to have typical SBP. While many
patients with this variant have cleared the bacterascites at the time of a
subsequent paracentesis, nearly 40% will develop typical SBP; thus follow-up
paracentesis is usually warranted in this setting.
![]() Treatment
Treatment
Empirical
therapy with cefotaxime or ampicillin and an aminoglycoside should be initiated
when the diagnosis is first suspected because enteric gram-negative bacilli are
found in the majority of cases; less frequently, the infection is caused by
pneumococci and other gram-positive bacteria. Cefotaxime is preferable due to
the lower rate of renal toxicity. Specific antibiotic therapy can be selected
once the specific organism is identified. Therapy is usually administered for
10 to 14 days, although one controlled study has suggested that a 5-day course
of intravenous antibiotics may be as effective when repeat paracentesis at 48 h
demonstrates a decline in the ascitic polymorphonuclear leukocytes count by
more than 50% and negative cultures.
While
appropriate antibiotic therapy is usually effective in the treatment of an
episode of SBP, recurrent episodes are relatively common; as many as 70% of
patients will experience at least one recurrence within a year of the first
episode. The risk of recurrence likely reflects the predisposing role of the
underlying advanced liver disease that contributed to the development of the
first episode of SBP. Recent trials have demonstrated that prophylactic
maintenance therapy with norfloxacin (400 mg/d) can reduce the frequency of
recurrent SBP. This agent presumably causes selective decontamination of the
intestine, eliminating many aerobic gram-negative bacilli.
Trimethoprim-sulfamethoxazole given for 5 days a week has also proven
effective. Antibiotics may be administered as infrequently as once a week
(e.g., ciprofoxacin, 750 mg once weekly). While maintenance therapy reduces the
frequency of SBP and need for hospitalization, it is unclear whether this is
associated with prolonged survival. Primary prevention of SBP in a subset of
high-risk cirrhotic patients [ascitic fluid protein <10 g/L (<1.0 g/dL)]
also appears to be warranted, as is prophylaxis for SBP during variceal
hemorrhage.
Hepatorenal Syndrome
Definition and Pathogenesis
Hepatorenal
syndrome is a serious complication in the patient with cirrhosis and ascites
and is characterized by worsening azotemia with avid sodium retention and
oliguria in the absence of identifiable specific causes of renal dysfunction.
The exact basis for this syndrome is not clear, but altered renal hemodynamics
appear to be involved. The kidneys are structurally intact; urinalysis and
pyelography are usually normal. Renal biopsy, although rarely needed, is also
normal, and in fact, kidneys from such patients have been used successfully for
renal transplantation. There are indications that an imbalance in certain
metabolites of arachidonic acid (prostaglandins and thromboxane) may play a
pathogenetic role.
Clinical Features and Diagnosis
Worsening
azotemia, hyponatremia, progressive oliguria, and hypotension are the hallmarks
of the hepatorenal syndrome. This syndrome, which is distinct from prerenal
azotemia, may be precipitated by severe gastrointestinal bleeding, sepsis, or
overly vigorous attempts at diuresis or paracentesis; it may also occur without
an obvious cause. It is essential to exclude other causes of renal impairment
often seen in these patients. These include prerenal azotemia or acute tubular necrosis
due to hypovolemia (e.g., secondary to gastrointestinal bleeding or diuretic
therapy) or an increased nitrogen load such as that seen as a result of
bleeding. Drug nephrotoxicity is also often a consideration, particularly in
the patient who has received agents such as aminoglycosides or contrast dye.
The diagnosis rests on the finding of an elevated serum creatinine level
[>133 ![]() mol/L
(>1.5 g/dL)] that fails to improve with volume expansion or withdrawal of
diuretics, together with an unremarkable urine sediment. The diagnosis is
supported by the demonstration of avid urinary sodium retention. Typically, the
urine sodium concentration is <5 mmol/L, a concentration lower than that
generally found in uncomplicated prerenal azotemia.
mol/L
(>1.5 g/dL)] that fails to improve with volume expansion or withdrawal of
diuretics, together with an unremarkable urine sediment. The diagnosis is
supported by the demonstration of avid urinary sodium retention. Typically, the
urine sodium concentration is <5 mmol/L, a concentration lower than that
generally found in uncomplicated prerenal azotemia.
![]() Treatment
Treatment
Treatment
is usually unsuccessful. Although some patients with hypotension and decreased
plasma volume may respond to infusions of salt-poor albumin, volume expansion
must be undertaken with caution to avoid precipitating variceal bleeding.
Vasodilator therapy, including intravenous infusions of low dose dopamine, is
not effective. While TIPS has been reported to improve renal function in some
patients, its use can not be recommended. In appropriate candidates, the
treatment of choice for hepatorenal syndrome is liver transplantation.
Hepatic Encephalopathy
Definition
Hepatic
(portal-systemic) encephalopathy is a complex neuropsychiatric syndrome
characterized by disturbances in consciousness and behavior, personality
changes, fluctuating neurologic signs, asterixis or "flapping
tremor," and distinctive electroencephalographic changes. Encephalopathy
may be acute and reversible or chronic and progressive. In severe
cases, irreversible coma and death may occur. Acute episodes may recur with
variable frequency.
Pathogenesis
The
specific cause of hepatic encephalopathy is unknown. The most important factors
in the pathogenesis are severe hepatocellular dysfunction and/or intrahepatic
and extrahepatic shunting of portal venous blood into the systemic circulation
so that the liver is largely bypassed. As a result of these processes, various
toxic substances absorbed from the intestine are not detoxified by the liver
and lead to metabolic abnormalities in the central nervous system (CNS). Ammonia
is the substance most often incriminated in the pathogenesis of encephalopathy.
Many, but not all, patients with hepatic encephalopathy have elevated blood
ammonia levels, and recovery from encephalopathy is often accompanied by
declining blood ammonia levels. Other compounds and metabolites that may
contribute to the development of encephalopathy include mercaptans (derived
from intestinal metabolism of methionine), short-chain fatty acids, and phenol.
False neurochemical transmitters (e.g., octopamine), resulting in part
from alterations in plasma levels of aromatic and branched-chain amino acids,
may also play a role. An increase in the permeability of the blood-brain
barrier to some of these substances may be an additional factor involved in the
pathogenesis of hepatic encephalopathy. Several observations suggest that
excessive concentrations of ![]() -aminobutyric
acid (GABA), an inhibitory neurotransmitter, in the CNS are important in the
reduced levels of consciousness seen in hepatic encephalopathy. Increased CNS
GABA may reflect failure of the liver to extract precursor amino acids
efficiently or to remove GABA produced in the intestine. In support of this,
there is also evidence to suggest that endogenous benzodiazepines, which act
through the GABA receptor, may contribute to the development of hepatic
encephalopathy. This evidence includes isolation of 1,4-benzodiazepines from
brain tissue of patients with fulminant hepatic failure as well as the partial
response observed in some patients and experimental animals after
administration of flumazenil, a benzodiazepine antagonist. However, the
inconsistent effect of flumazenil in patients with encephalopathy, as well as
potential methodologic pitfalls in the measurement of endogenous
benzodiazepines, preclude definitive attribution of a role to these substances
in the pathogenesis of hepatic encephalopathy. The finding of direct
enhancement of GABA receptor activation by ammonia suggests that several of the
factors described above may be operating via a final common pathway to produce
the neuronal depression of hepatic encephalopathy. Finally, the observation of
hyperintensity in the basal ganglia by magnetic resonance imaging in cirrhotic
patients suggests that excessive manganese deposition may also
contribute to the pathogenesis of hepatic encephalopathy. Further studies are
needed to determine whether chelation therapy exerts long-term benefit.
-aminobutyric
acid (GABA), an inhibitory neurotransmitter, in the CNS are important in the
reduced levels of consciousness seen in hepatic encephalopathy. Increased CNS
GABA may reflect failure of the liver to extract precursor amino acids
efficiently or to remove GABA produced in the intestine. In support of this,
there is also evidence to suggest that endogenous benzodiazepines, which act
through the GABA receptor, may contribute to the development of hepatic
encephalopathy. This evidence includes isolation of 1,4-benzodiazepines from
brain tissue of patients with fulminant hepatic failure as well as the partial
response observed in some patients and experimental animals after
administration of flumazenil, a benzodiazepine antagonist. However, the
inconsistent effect of flumazenil in patients with encephalopathy, as well as
potential methodologic pitfalls in the measurement of endogenous
benzodiazepines, preclude definitive attribution of a role to these substances
in the pathogenesis of hepatic encephalopathy. The finding of direct
enhancement of GABA receptor activation by ammonia suggests that several of the
factors described above may be operating via a final common pathway to produce
the neuronal depression of hepatic encephalopathy. Finally, the observation of
hyperintensity in the basal ganglia by magnetic resonance imaging in cirrhotic
patients suggests that excessive manganese deposition may also
contribute to the pathogenesis of hepatic encephalopathy. Further studies are
needed to determine whether chelation therapy exerts long-term benefit.
In
the patient with otherwise stable cirrhosis, hepatic encephalopathy often
follows a clearly identifiable precipitating event (Table 299-3). Perhaps the
most common predisposing factor is gastrointestinal bleeding, which
leads to an increase in the production of ammonia and other nitrogenous
substances, which are then absorbed. Similarly, increased dietary protein
may precipitate encephalopathy as a result of increased production of
nitrogenous substances by colonic bacteria. Electrolyte disturbances,
particularly hypokalemic alkalosis secondary to overzealous use of diuretics,
vigorous paracentesis, or vomiting, may precipitate hepatic encephalopathy.
Systemic alkalosis causes an increase in the amount of nonionic ammonia (NH3)
relative to ammonium ions NH4+). Only nonionic
(uncharged) ammonia readily crosses the blood-brain barrier and accumulates in
the CNS. Hypokalemia also directly stimulates renal ammonia production.
Injudicious use of CNS-depressing drugs (e.g., barbiturates, benzodiazepines)
and acute infection may trigger or aggravate hepatic encephalopathy, although
the mechanisms involved are not clear. Other potential precipitating factors
include superimposed acute viral hepatitis, alcoholic hepatitis, extrahepatic
bile duct obstruction, constipation, surgery, and other coincidental medical
complications.
Table 299-3: Common Precipitants of Hepatic
Encephalopathy
|
Hepatic
encephalopathy has protean manifestations, and any neurologic abnormality,
including focal deficits, may be encountered. In patients with acute
encephalopathy, neurologic deficits are completely reversible upon correction
of underlying precipitating factors and/or improvement in liver function, but
in patients with chronic encephalopathy, the deficits may be irreversible and
progressive. Cerebral edema is frequently present and contributes to the
clinical picture and overall mortality in patients with both acute and chronic
encephalopathy.
The
diagnosis of hepatic encephalopathy should be considered when four major
factors are present: (1) acute or chronic hepatocellular disease and/or
extensive portal-systemic collateral shunts (the latter may be either
spontaneous, e.g., secondary to portal hypertension, or surgically created,
e.g., portacaval anastomosis); (2) disturbances of awareness and mentation,
which may progress from forgetfulness and confusion to stupor and finally coma;
(3) shifting combinations of neurologic signs, including asterixis, rigidity,
hyperreflexia, extensor plantar signs, and rarely, seizures; and (4) a
characteristic (but nonspecific) symmetric, high-voltage, triphasic slow-wave
(2 to 5 per second) pattern on the electroencephalogram. Asterixis ("liver
flap," "flapping tremor") is a nonrhythmic asymmetric lapse in
voluntary sustained position of the extremities, head, and trunk. It is best
demonstrated by having the patient extend the arms and dorsiflex the hands.
Because elicitation of asterixis depends on sustained voluntary muscle
contraction, it is not present in the comatose patient. Asterixis is
nonspecific and also occurs in patients with other forms of metabolic brain
disease. Disturbances of sleep with reversal of sleep/wake cycles are among the
earliest signs of encephalopathy. Alterations in personality, mood
disturbances, confusion, deterioration in self-care and handwriting, and
daytime somnolence are additional clinical features of encephalopathy. Fetor
hepaticus, a unique musty odor of the breath and urine believed to be due
to mercaptans, may be noted in patients with varying stages of hepatic
encephalopathy.
Grading
or classifying the stages of hepatic encephalopathy is often helpful in
following the course of the illness and assessing response to therapy. One
useful classification is shown in Table 299-4.
Table 299-4: Clinical Stages of Hepatic
Encephalopathy
|
The
diagnosis of hepatic encephalopathy is usually one of exclusion. There are no
diagnostic liver function test abnormalities, although an elevated serum
ammonia level in the appropriate clinical setting is highly suggestive of the
diagnosis. Examination of the cerebrospinal fluid is unremarkable, and computed
tomography of the brain shows no characteristic abnormalities until late in
stage IV when cerebral edema may supervene. A number of conditions,
particularly disorders related to acute and chronic alcoholism, can mimic the
clinical features of hepatic encephalopathy. These include acute alcohol
intoxication, sedative overdose, delirium tremens, Wernicke's encephalopathy,
and Korsakoff's psychosis (Chap. 373). Subdural hematoma, meningitis, and
hypoglycemia or other metabolic encephalopathies must also be considered,
especially in patients with alcoholic cirrhosis. In young patients with liver
disease and neurologic abnormalities, Wilson's disease should be excluded.
![]() Treatment
Treatment
(See
Fig. 299-4) Early recognition and prompt treatment of hepatic encephalopathy
are essential. Patients with acute, severe hepatic encephalopathy (stage IV)
require the usual supportive measures for the comatose patient. Specific
treatment of hepatic encephalopathy is aimed at (1) elimination or treatment of
precipitating factors and (2) lowering of blood ammonia (and other toxin)
levels by decreasing the absorption of protein and nitrogenous products from
the intestine. In the setting of acute gastrointestinal bleeding, blood in the
bowel should be promptly evacuated with laxatives (and enemas if necessary) in
order to reduce the nitrogen load. Protein should be excluded from the diet,
and constipation should be avoided. Ammonia absorption can be decreased by the
administration of lactulose, a nonabsorbable disaccharide that acts as an
osmotic laxative. Metabolism of lactulose by colonic bacteria may also result
in an acid pH that favors conversion of ammonia to the poorly absorbed ammonium
ion. In addition, lactulose may actually diminish ammonia production through
its direct effects on bacterial metabolism. Acutely, lactulose syrup can be
administered in a dose of 30 to 60 mL every hour until diarrhea occurs;
thereafter the dose is adjusted (usually 15 to 30 mL three times daily) so that
the patient has two to four soft stools daily. Intestinal ammonia production by
bacteria can also be decreased by oral administration of a
"nonabsorbable" antibiotic such as neomycin (0.5 to 1.0 g every 6 h).
However, despite poor absorption, neomycin may reach sufficient concentrations
in the bloodstream to cause renal toxicity. Equal benefits may be achieved with
broad-spectrum antibiotics such as metronidazole. The use of agents such as
levodopa, bromocriptine, keto analogues of essential amino acids, and
intravenous amino acid formulations rich in branched-chain amino acids in the
treatment of acute hepatic encephalopathy remains of unproven benefit.
Flumazenil, a short-acting benzodiazepine antagonist, may have a role in
management of hepatic encephalopathy precipitated by use of benzodiazepines, if
there is a need for urgent therapy. Hemoperfusion to remove toxic substances
and therapy directed primarily toward coincident cerebral edema in acute
encephalopathy are also of unproven value. The efficacy of extracorporeal liver
assist devices employing hepatocytes of porcine or human origin to bridge
patients to recovery or transplantation is as yet unproven but is currently
being studied.
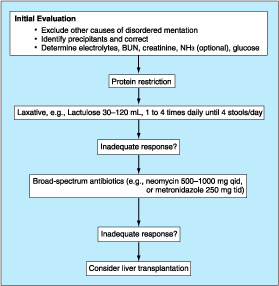
Figure 299-4: Approach to the patient with hepatic
encephalopathy. BUN, blood urea nitrogen.
Chronic
encephalopathy may be effectively controlled by administration of lactulose.
Management of patients with chronic encephalopathy should include dietary
protein restriction (usually to 60 g/d) in combination with low doses of
lactulose or neomycin. Nephrotoxicity or ototoxicity may be limiting in
prolonged usage of neomycin. There are suggestions that vegetable protein may
be preferable to animal protein.
Other Sequelae of Cirrhosis
Coagulopathy
Patients
with cirrhosis often demonstrate a variety of abnormalities in both cellular
and humoral clotting function. Thrombocytopenia may result from hypersplenism.
In the alcoholic patient, there may be direct bone marrow suppression by
ethanol. Diminished protein synthesis may lead to reduced production of
fibrinogen (factor I), prothrombin (factor II), and factors V, VII, IX, and X.
Reduction in levels of all factors except factor V may be worsened by the
coincident malabsorption of the fat-soluble cofactor vitamin K due to
cholestasis (Chap. 286). Of these, factor VII appears to be pivotal. In
cirrhosis, it is the first of the factors to become depleted and, because of
its short half-life, replacement with plasma often fails to correct an elevated
prothrombin time. Preliminary studies suggest that selective replacement of
factor VII can correct the prothrombin time in patients with cirrhosis.
Hepatocellular Carcinoma
Hypoxemia and Hepatopulmonary Syndrome
Definition and Pathogenesis
Mild
hypoxemia occurs in approximately one-third of patients with chronic liver
disease. The hepatopulmonary syndrome is typically manifest by hypoxemia,
platypnea, and orthodeoxia. Hypoxemia usually results from right-to-left
intrapulmonary shunts through dilatations in intrapulmonary vessels that can be
detected by contrast-enhanced echocardiography or a macroaggregated albumin
lung perfusion scan. The mechanisms of shunt formation are unclear, but one
animal model suggests that endothelin-1 levels and pulmonary nitric oxide,
raised in cirrhosis, correlate with degree of shunting.
![]() Treatment
Treatment
No
specific treatment is consistently effective, though large arteriovenous shunts
may be embolized. It is now increasingly recognized that liver transplantation
may eventually lead to amelioration of the hepatopulmonary syndrome in cases
that have not yet been complicated by advanced pulmonary hypertension.