Diabetes Mellitus
Diabetes
mellitus (DM) comprises a group of common metabolic disorders that share the
phenotype of hyperglycemia. Several distinct types of DM exist and are caused
by a complex interaction of genetics, environmental factors, and life-style
choices. Depending on the etiology of the DM, factors contributing to
hyperglycemia may include reduced insulin secretion, decreased glucose usage,
and increased glucose production. The metabolic dysregulation associated with
DM causes secondary pathophysiologic changes in multiple organ systems that
impose a tremendous burden on the individual with diabetes and on the health
care system. In the United States, DM is the leading cause of end-stage renal
disease, nontraumatic lower extremity amputations, and adult blindness. With an
increasing incidence worldwide, DM will likely continue to be a leading cause
of morbidity and mortality for the foreseeable future.
CLASSIFICATION
Recent
advances in the understanding of the etiology and pathogenesis of diabetes have
led to a revised classification (Table 333-1). Although all forms of DM are
characterized by hyperglycemia, the pathogenic mechanisms by which
hyperglycemia arises differ widely. Some forms of DM are characterized by an
absolute insulin deficiency or a genetic defect leading to defective insulin
secretion, whereas other forms share insulin resistance as their underlying
etiology. Recent changes in classification reflect an effort to classify DM on
the basis of the pathogenic process that leads to hyperglycemia, as opposed to
criteria such as age of onset or type of therapy (Fig. 333-1).
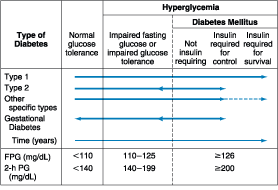
Figure 333-1: Spectrum of glucose homeostasis and
diabetes. The spectrum from normal glucose tolerance to diabetes in type 1
diabetes, type 2 diabetes, other specific types of diabetes, and gestational
diabetes is shown from left to right. In most types of diabetes, the individual
traverses from normal glucose tolerance to impaired glucose tolerance to frank
diabetes. Arrows indicate that changes in glucose tolerance may be
bi-directional in some types of diabetes. For example, individuals with type 2
diabetes may return to the impaired glucose tolerance category with weight
loss; in gestational diabetes, diabetes may revert to impaired glucose
tolerance or even normal glucose tolerance after delivery. The fasting plasma
glucose (FPG) and 2-h plasma glucose (PG), after a glucose challenge for the different
categories of glucose tolerance, are shown at the lower part of the figure (as
defined by recent consensus panels-see text). These values do not apply to the
diagnosis of gestational diabetes. Some types of diabetes may or may not
require insulin for survival, hence the dotted line. (Conventional units are
used in the figure.)(Adapted from American Diabetes Association: Clinical
Practice Guidelines, 2000)
Table 333-1: Etiologic Classification of
Diabetes Mellitus
|
The
two broad categories of DM are designated type 1 and type 2. Type 1A DM results
from autoimmune beta cell destruction, which usually leads to insulin
deficiency. Type 1B DM is also characterized by insulin deficiency as well as a
tendency to develop ketosis. However, individuals with type 1B DM lack
immunologic markers indicative of an autoimmune destructive process of the beta
cells. The mechanisms leading to beta cell destruction in these patients are
unknown. Relatively few patients with type 1 DM fall into the type 1B
idiopathic category; many of these individuals are either African-American or
Asian in heritage.
Type
2 DM is a heterogeneous group of disorders usually characterized by variable
degrees of insulin resistance, impaired insulin secretion, and increased
glucose production. Distinct genetic and metabolic defects in insulin action
and/or secretion give rise to the common phenotype of hyperglycemia in type 2
DM (see below). The identification of distinct pathogenic processes in type 2
DM has important potential therapeutic implications, as pharmacologic agents
that target specific metabolic derangements become available.
Two
features of the current classification of DM diverge from previous
classifications. First, the terms insulin-dependent diabetes mellitus
(IDDM) and noninsulin-dependent diabetes mellitus (NIDDM) are obsolete.
These previous designations reflected the observation that most individuals
with type 1 DM (previously IDDM) have an absolute requirement for insulin
treatment, whereas many individuals with type 2 DM (previously NIDDM) do not
require insulin therapy to prevent ketoacidosis. However, because many
individuals with type 2 DM eventually require insulin treatment for control of
glycemia, the use of the latter term generated considerable confusion.
A
second difference is that age is no longer used as a criterion in the new
classification system. Although type 1 DM most commonly develops before the age
of 30, an autoimmune beta cell destructive process can develop at any age. In
fact, it is estimated that between 5 and 10% of individuals who develop DM
after age 30 have type 1A DM. Likewise, although type 2 DM more typically
develops with increasing age, it also occurs in children, particularly in obese
adolescents.
OTHER TYPES OF DM
Other
etiologies for DM include specific genetic defects in insulin secretion or
action, metabolic abnormalities that impair insulin secretion, and a host of
conditions that impair glucose tolerance (Table 333-1). Maturity onset
diabetes of the young (MODY) is a subtype of DM characterized by autosomal
dominant inheritance, early onset of hyperglycemia, and impairment in insulin
secretion (discussed below). Mutations in the insulin receptor cause a group of
rare disorders characterized by severe insulin resistance.
DM
can result from pancreatic exocrine disease when the majority of pancreatic
islets (>80%) are destroyed. Several endocrinopathies can lead to DM as a
result of excessive secretion of hormones that antagonize the action of
insulin. Notable within this group are acromegaly and Cushing's disease, both
of which may present with DM. Viral infections have been implicated in
pancreatic islet destruction, but are an extremely rare cause of DM. Congenital
rubella greatly increases the risk for DM; however, most of these individuals
also have immunologic markers indicative of autoimmune beta cell destruction.
GESTATIONAL DIABETES MELLITUS (GDM)
Glucose
intolerance may develop and first become recognized during pregnancy. Insulin
resistance related to the metabolic changes of late pregnancy increases insulin
requirements and may lead to hyperglycemia or impaired glucose tolerance. GDM
is seen in approximately 4% of pregnancies in the United States; most women
revert to normal glucose tolerance post-partum but have a substantial risk (30
to 60%) of developing DM later in life
EPIDEMIOLOGY
The
worldwide prevalence of DM has risen dramatically over the past two decades. It
is projected that the number of individuals with DM will continue to increase
in the near future. Between 1976 and 1994, for example, the prevalence of DM
among adults in the United States increased from 8.9% to 12.3%. These findings,
based on national epidemiologic data, include individuals with a diagnosis of
DM and those with undiagnosed DM (based on identical diagnostic criteria).
Likewise, prevalence rates of impaired fasting glucose (IFG) increased from
6.5% to 9.7% over the same period. Although the prevalence of both type 1 and
type 2 DM is increasing worldwide, the prevalence of type 2 DM is expected to
rise more rapidly in the future because of increasing obesity and reduced
activity levels.
There
is considerable geographic variation in the incidence of both type 1 and type 2
DM. For example, Scandinavia has the highest rate of type 1 DM (in Finland,
incidence is 35/100,000 per year). The Pacific Rim has a much lower rate (in
Japan and China, incidence is 1 to 3/100,000 per year) of type 1 DM; Northern
Europe and the United States share an intermediate rate (8 to 17/100,000 per
year). Much of the increased risk of type 1 DM is believed to reflect the
frequency of high-risk HLA alleles among ethnic groups in different geographic
locations.
The
prevalence of type 2 DM and its harbinger, impaired glucose tolerance (IGT), is
highest in certain Pacific islands, intermediate in countries such as India and
the United States, and relatively low in Russia and China. This variability is
likely due to both genetic and environmental factors. There is also
considerable variation in DM prevalence among different ethnic populations
within a given country.
In
1998, approximately 16 million individuals in the United States met the
diagnostic criteria for DM. This represents ~6% of the population. About
800,000 individuals in the United States develop DM each year. The vast
majority of these (>90%) have type 2 DM. The number of people with DM
increases with the age of the population, ranging from an incidence of ~1.5% in
individuals from 20 to 39 years to ~20% of individuals >75 years. The
incidence of DM is similar in men and women throughout most age ranges but is
slightly greater in men >60 years. The prevalence of DM is approximately
twofold greater in African Americans, Hispanic Americans, and Native Americans
than in non-Hispanic whites, and the onset of type 2 DM occurs, on average, at
an earlier age in the former groups than in the non-Hispanic white population.
The incidence of type 2 DM in these ethnic groups is rapidly increasing. The
reasons for these differences are not yet clear.
DIAGNOSIS
Revised
criteria for diagnosing DM have been issued by consensus panels of experts from
the National Diabetes Data Group and the World Health Organization (Table
333-2). The revised criteria reflect new epidemiologic and metabolic evidence
and are based on the following premises: (1) the spectrum of fasting plasma
glucose (FPG) and the response to an oral glucose load varies in normal
individuals, and (2) DM defined as the level of glycemia at which
diabetes-specific complications are noted and not on the level of glucose
tolerance from a population-based viewpoint. For example, the prevalence of
retinopathy in Native Americans (Pima Indian population) begins to increase at
a FPG > 6.4 mmol/L (116 mg/dL) (Fig. 333-2).
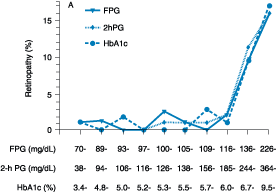
Figure 333-2: Relationship of diabetes-specific
complication and glucose tolerance. This figure shows the incidence of
retinopathy in Pima Indians as a function of the fasting plasma glucose (FPG),
the 2-h plasma glucose after a 75-g oral glucose challenge (2hPG), or glycated
hemoglobin (HbA1c). Note that the incidence of retinopathy greatly increases at
a fasting plasma glucose >116 mg/dL, or a 2-h plasma glucose of 185 mg/dL,
or a HbA1c >6.0%. (Conventional units are used in the figure.)(From American
Diabetes Association: Clinical Practice Guidelines, 2000, as adapted from
McCance et al: BMJ 308:1323, 1994)
Table 333-2: Criteria for the Diagnosis of
Diabetes Mellitus
|
|||||||||
|
|
||||||||
Glucose
tolerance is classified into three categories based on the FPG: (1) FPG <
6.1 mmol/L (110 mg/dL) is considered normal; (2) FPG ![]() 6.1
mmol/L (110 mg/dL) but < 7.0 mmol/L (126 mg/dL) is defined as IFG; and (3)
FPG
6.1
mmol/L (110 mg/dL) but < 7.0 mmol/L (126 mg/dL) is defined as IFG; and (3)
FPG ![]() 7.0
mmol/L (126 mg/dL) warrants the diagnosis of DM. IFG is a new diagnostic
category defined by the Expert Committee on the Diagnosis and Classification of
Diabetes Mellitus. It is analogous to IGT, which is defined as plasma glucose
levels between 7.8 and 11.1 mmol/L (140 and 200 mg/dL) 2 h after a 75-g oral
glucose load (Table 333-2). Individuals with IFG or IGT are at substantial risk
for developing type 2 DM and cardiovascular disease in the future, though they
may not meet the criteria for DM.
7.0
mmol/L (126 mg/dL) warrants the diagnosis of DM. IFG is a new diagnostic
category defined by the Expert Committee on the Diagnosis and Classification of
Diabetes Mellitus. It is analogous to IGT, which is defined as plasma glucose
levels between 7.8 and 11.1 mmol/L (140 and 200 mg/dL) 2 h after a 75-g oral
glucose load (Table 333-2). Individuals with IFG or IGT are at substantial risk
for developing type 2 DM and cardiovascular disease in the future, though they
may not meet the criteria for DM.
The
revised criteria for the diagnosis of DM emphasize the FPG as the most reliable
and convenient test for diagnosing DM in asymptomatic individuals. A random
plasma glucose concentration ![]() 11.1
mmol/L (200 mg/dL) accompanied by classic symptoms of DM (polyuria, polydipsia,
weight loss) is sufficient for the diagnosis of DM (Table 333-2). Oral glucose
tolerance testing, although still a valid mechanism for diagnosing DM, is not
recommended as part of routine screening.
11.1
mmol/L (200 mg/dL) accompanied by classic symptoms of DM (polyuria, polydipsia,
weight loss) is sufficient for the diagnosis of DM (Table 333-2). Oral glucose
tolerance testing, although still a valid mechanism for diagnosing DM, is not
recommended as part of routine screening.
Some
investigators have advocated the hemoglobin A1c (HbA1c) as a diagnostic test
for DM. Though there is a strong correlation between elevations in the plasma
glucose and the HbA1c (discussed below), the relationship between the FPG and
the HbA1c in individuals with normal glucose tolerance or mild glucose
intolerance is less clear, and the test is not universally standardized or
available.
The
diagnosis of DM has profound implications for an individual from both a medical
and financial standpoint. Thus, the health care provider must be certain that
these criteria are completely satisfied before assigning the diagnosis of DM to
an individual. The revised criteria also allow for the diagnosis of DM to be
withdrawn in situations where the FPG no longer exceeds these criteria.
Abnormalities on screening tests for diabetes should be repeated before making
a definitive diagnosis of DM, unless acute metabolic derangements or a markedly
elevated plasma glucose are present (Table 333-2).
SCREENING
Widespread
use of the FPG as a screening test for type 2 DM is strongly encouraged
because: (1) a large number of individuals who meet the current criteria for DM
are unaware that they have the disorder, (2) epidemiologic studies suggest that
type 2 DM may be present for up to a decade before diagnosis, and (3) as many
as 50% of individuals with type 2 DM have one or more diabetes-specific
complications at the time of their diagnosis. The Expert Committee suggests
screening all individuals >45 years every 3 years and screening asymptomatic
individuals with additional risk factors (Table 333-3) at an earlier age. In
contrast to type 2 DM, it is rare for an individual to have a long asymptomatic
period of hyperglycemia prior to the diagnosis of type 1 DM. A number of
immunologic markers for type 1 DM are becoming available (discussed below), but
their use is currently discouraged pending the identification of clinically
beneficial interventions for individuals at high risk for developing type 1 DM.
Table 333-3: Risk Factors for Type 2 Diabetes
Mellitus
|
||||
|
|
|||
INSULIN BIOSYNTHESIS, SECRETION, AND ACTION
BIOSYNTHESIS
Insulin
is produced in the beta cells of the pancreatic islets. It is initially
synthesized as a single-chain 86-amino-acid precursor polypeptide,
preproinsulin. Subsequent proteolytic processing removes the aminoterminal
signal peptide, giving rise to proinsulin. Proinsulin is structurally related
to insulin-like growth factors I and II, which bind weakly to the insulin
receptor (Chap. 327). Cleavage of an internal 31-residue fragment from
proinsulin generates the C peptide and the A (21 amino acids) and B (30 amino
acids) chains of insulin, which are connected by disulfide bonds. The mature
insulin molecule and C peptide are stored together and cosecreted from
secretory granules in the beta cells. Because the C peptide is less susceptible
than insulin to hepatic degradation, it is a useful a marker of insulin
secretion and allows discrimination of endogenous and exogenous sources of
insulin in the evaluation of hypoglycemia (Chap. 334). Human insulin is now
produced by recombinant DNA technology; structural alterations at one or more
residues are useful for modifying its physical and pharmacologic
characteristics (see below).
SECRETION
Glucose
is the key regulator of insulin secretion by the pancreatic beta cell, although
amino acids, ketones, various nutrients, gastrointestinal peptides, and
neurotransmitters also influence insulin secretion. Glucose levels >3.9
mmol/L (70 mg/dL) stimulate insulin synthesis, primarily by enhancing protein
translation and processing, as well as inducing insulin secretion. Glucose
stimulates insulin secretion through a series of regulatory steps that begin
with transport into the beta cell by the GLUT2 glucose transporter (Fig.
333-3). Glucose phosphorylation by glucokinase is the rate-limiting step that
controls glucose-regulated insulin secretion.
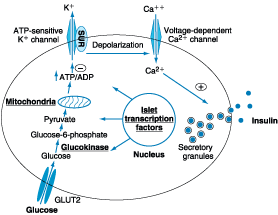
Figure 333-3: Diabetes and abnormalities in
glucose-stimulated insulin secretion. Glucose and other nutrients regulate
insulin secretion by the pancreatic beta cell. Glucose is transported by the
GLUT2 glucose transporter; subsequent glucose metabolism by the beta cell
alters ion channel activity, leading to insulin secretion. The SUR receptor is
the binding site for oral hypoglycemic agents. Mutations in the events or
proteins underlined are a cause of maturity onset diabetes of the young (MODY)
or other forms of diabetes. SUR, sulfonylurea receptor; ATP, adenosine
triphosphate; ADP, adenosine diphosphate.(Adapted from Lowe, 1998.)
Further
metabolism of glucose-6-phosphate via glycolysis generates ATP, which inhibits
the activity of an ATP-sensitive K+ channel. This channel is a
complex of two separate proteins, one of which is the receptor for certain oral
hypoglycemics (e.g., sulfonylureas, meglitinides); the other subunit is an
inwardly rectifying K+ channel protein. Inhibition of this K+
channel induces beta cell membrane depolarization, opening of voltage-dependent
calcium channels (leading to an influx of calcium), and stimulation of insulin
secretion. Careful studies of insulin secretory profiles reveal pulsatile pattern
of hormone release, with small secretory bursts occurring about every 10 min,
superimposed upon greater amplitude oscillations of about 80 to 150 min. Meals
or other major stimuli of insulin secretion induce large (four- to fivefold
increase versus baseline) bursts of insulin secretion that usually last for 2
to 3 h before returning to baseline. Derangements in these normal secretory
patterns are one of the earliest signs of beta cell dysfunction in DM (see
below).
ACTION
Once
insulin is secreted into the portal vein, ~50% is removed and degraded by the
liver. Unextracted insulin enters the systemic circulation and binds to its
receptor in target sites. The insulin receptor belongs to the tyrosine kinase
class of membrane-bound receptors (Chap. 327). Insulin binding to the receptor
stimulates intrinsic tyrosine kinase activity, leading to receptor
autophosphorylation and the recruitment of intracellular signaling molecules,
such as insulin receptor substrates (IRS) 1 and 2 (Fig. 333-4). These and other
adaptor proteins initiate a complex cascade of phosphorylation and
dephosphorylation reactions, ultimately resulting in the widespread metabolic
and mitogenic effects of insulin. As an example, activation of the
phosphatidylinositol-3′-kinase (PI-3 kinase) pathway stimulates
translocation of glucose transporters (e.g., GLUT4) to the cell surface, an
event that is crucial for glucose uptake by skeletal muscle and fat. Activation
of other insulin receptor signaling pathways induces glycogen synthesis,
protein synthesis, lipogenesis, and regulation of various genes in
insulin-responsive cells.
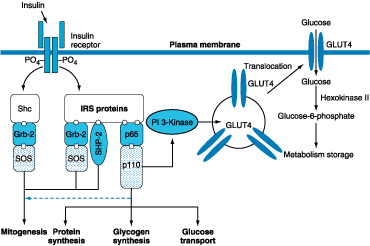
Figure 333-4: Insulin signal transduction pathway. The
insulin receptor has intrinsic tyrosine kinase activity and interacts with
insulin receptor substrates (IRS and Shc) proteins. A number of
"docking" proteins bind to these cellular proteins and initiate the
metabolic actions of insulin [GrB-2, SOS, SHP-2, p65, p110, and phosphoinositol
phosphate 3-kinase (PI 3-kinase)]. Insulin increases glucose transport through
PI 3-kinase, which promotes the translocation of intracellular vesicles
containing GLUT4 glucose transporter to the plasma membrane.(Adapted from Lowe,
1998; Virkamaki et al, 1999)
Glucose
homeostasis reflects a precise balance between hepatic glucose production and
peripheral glucose uptake and utilization. Insulin is the most important
regulator of this metabolic equilibrium, but the effects of other pathways
including neural input, metabolic signals, and hormones (e.g., glucagon) result
in integrated control of glucose supply and utilization (Chap. 334 and 334-1).
In the fasting state, low insulin levels promote hepatic gluconeogenesis and
glycogenolysis to prevent hypoglycemia. Low insulin levels decrease glycogen
synthesis, reduce glucose uptake in insulin-sensitive tissues, and promote
mobilization of stored precursors. Reduced insulin levels are also permissive
in allowing glucagon to stimulate glycogenolysis and gluconeogenesis by the
liver and renal medulla. These processes are of critical importance to ensure
an adequate glucose supply for the brain. Postprandially, a large glucose load
elicits a rise in insulin and fall in glucagon, leading to a reversal of these
processes. The major portion of postprandial glucose is utilized by skeletal
muscle. Other tissues, most notably the brain, utilize glucose in an
insulin-independent fashion.
PATHOGENESIS
TYPE 1 DM
Type
1A DM develops as a result of the synergistic effects of genetic,
environmental, and immunologic factors that ultimately destroy the pancreatic
beta cells. The temporal development of type 1A DM is shown schematically as a
function of beta cell mass in Fig. 333-5. Individuals with a genetic
susceptibility have normal beta cell mass at birth but begin to lose beta cells
secondary to autoimmune destruction that occurs over months to years. This
autoimmune process is thought to be triggered by an infectious or environmental
stimulus and to be sustained by a beta cell-specific molecule. In the majority
of individuals, immunologic markers appear after the triggering event but
before diabetes becomes clinically overt. Beta cell mass then begins to
decline, and insulin secretion becomes progressively impaired, although normal
glucose tolerance is maintained. The rate of decline in beta cell mass varies
widely among individuals, with some patients progressing rapidly to clinical
diabetes and others evolving more slowly. Features of diabetes do not become
evident until a majority of beta cells are destroyed (~80%). At this point,
residual functional beta cells still exist but are insufficient in number to
maintain glucose tolerance. The events that trigger the transition from glucose
intolerance to frank diabetes are often associated with increased insulin
requirements, as might occur during infections or puberty. Following the
initial clinical presentation of type 1A DM, a "honeymoon" phase may
ensue during which time glycemic control is achieved with modest doses of
insulin or, rarely, insulin is not needed. However, this fleeting phase of
endogenous insulin production from residual beta cells disappears as the
autoimmune process destroys the remaining beta cells, and the individual becomes
completely insulin deficient.
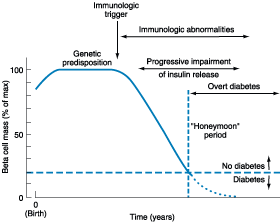
Figure 333-5: Temporal model for development of type 1
diabetes. Individuals with a genetic predisposition are exposed to an
immunologic trigger that initiates an autoimmune process, resulting in a
gradual decline in beta cell mass. The downward slope of the beta cell mass
varies among individuals. This progressive impairment in insulin release
results in diabetes when ~80% of the beta cell mass is destroyed. A
"honeymoon" phase may be seen in the first 1 or 2 years after the
onset of diabetes and is associated with reduced insulin requirements. (Adapted
from Medical Management of Type 1 Diabetes, 3d ed, JS Skyler (ed). Alexandria,
VA, American Diabetes Association, 1998)
![]() Genetics
Genetics
The
genetic contributions to type 1A DM involve multiple genes. The development of
the disease appears to require inheritance of a sufficient complement of genes
to confer susceptibility to the disorder. The concordance of type 1A DM in
identical twins ranges between 30 and 70%, indicating that additional modifying
factors must be involved in determining whether diabetes develops. The major
susceptibility gene for type 1A DM is located in the HLA region on chromosome
6. Polymorphisms in the HLA complex appear to account for 40 to 50% of the
genetic risk of developing type 1A DM. This region contains genes that encode
the class II MHC molecules, which present antigen to helper T cells and thus
are involved in initiating the immune response (Chaps. 305, 306 and 307). The
ability of class II MHC molecules to present antigen is dependent on the amino
acid composition of their antigen-binding sites. Amino acid substitutions may
influence the specificity of the immune response by altering the binding
affinity of different antigens for the class II molecules.
Most
individuals with type 1A DM have the HLA DR3 and/or DR4 haplotype. Refinements
in genotyping of HLA loci have shown that the haplotypes DQA1*0301, DQB1*0302
and DQA1*501, DQB1*0201 have the strongest association with type 1A DM. These
haplotypes are present in 40% of children with type 1A DM as compared to 2% of
the normal U.S. population.
In
addition to MHC class II associations, at least 17 different genetic loci may
contribute susceptibility to type 1A DM. For example, polymorphisms in the
promoter region of the insulin gene appear to account for ~10% of the
predisposition to type 1A DM. Genes that confer protection against the
development of the disease also exist. For example, the haplotype DQA1*0102,
DQB1*0602 is present in 20% of the U.S. population but is extremely rare in
individuals with type 1A DM (<1%).
Although
type 1A DM is clearly associated with certain predisposing genotypes, most
individuals with these haplotypes do not develop diabetes. In addition, most
individuals with type 1A DM do not have a first-degree relative with this
disorder. Nevertheless, the risk of developing type 1A DM for relatives of
individuals with the disease is considerably higher compared to the risk for
the general population.
Autoimmune Factors
Although
other islet cell types [alpha cells (glucagon-producing), delta cells
(somatostatin-producing) or PP cells (pancreatic polypeptide-producing)] are
functionally and embryologically similar to beta cells and express most of the
same proteins as beta cells, they are inexplicably spared from the autoimmune
process. Pathologically, the pancreatic islets are infiltrated with lymphocytes
(in a process termed insulitis). After all beta cells are destroyed, the
inflammatory process abates, the islets become atrophic, and immunologic markers
disappear. Studies of the insulitis and autoimmune process in humans and animal
models of type 1A DM (NOD mouse and BB rat) have identified the following
abnormalities in both the humoral and cellular arms of the immune system: (1)
islet cell autoantibodies; (2) activated lymphocytes in the islets,
peripancreatic lymph nodes, and systemic circulation; (3) T lymphocytes that
proliferate when stimulated with islet proteins; and (4) release of cytokines
within the insulitis. Beta cells seem to be particularly susceptible to the
toxic effect of some cytokines (tumor necrosis factor ![]() ,
interferon
,
interferon ![]() ,
and interleukin 1). The precise mechanisms of beta cell death are not known but
may involve formation of nitric oxide metabolites, apoptosis, and direct CD8+ T
cell cytotoxicity. Islet autoantibodies are not thought to be involved in the
destructive process, as these antibodies do not generally react with the cell
surface of islet cells and are not capable of transferring diabetes mellitus to
animals.
,
and interleukin 1). The precise mechanisms of beta cell death are not known but
may involve formation of nitric oxide metabolites, apoptosis, and direct CD8+ T
cell cytotoxicity. Islet autoantibodies are not thought to be involved in the
destructive process, as these antibodies do not generally react with the cell
surface of islet cells and are not capable of transferring diabetes mellitus to
animals.
Pancreatic
islet molecules targeted by the autoimmune process include insulin, glutamic
acid decarboxylase (GAD; the biosynthetic enzyme for the neurotransmitter
GABA), ICA-512/IA-2 (homology with tyrosine phosphatases), and phogrin (insulin
secretory granule protein). Other less clearly defined autoantigens include an
islet ganglioside and carboxypeptidase H. With the exception of insulin, none
of the autoantigens are beta cell specific, which raises the question of how
the beta cells are selectively destroyed. Current theories favor initiation of
an autoimmune process directed at one beta cell molecule, which then spreads to
other islet molecules as the immune process destroys beta cells and creates a
series of secondary autoantigens. The beta cells of individuals who develop
type 1A DM do not differ from beta cells of normal individuals, since
transplanted islets are destroyed by a recurrence of the autoimmune process of
type 1A DM.
Immunologic Markers
Islet
cell autoantibodies (ICAs) are a composite of several different antibodies
directed at pancreatic islet molecules such as GAD, insulin, IA-2/ICA512, and
an islet ganglioside and serve as a marker of the autoimmune process of type 1A
DM. Testing for ICAs can be useful in classifying the type of DM as type IA and
in identifying nondiabetic individuals at risk for developing type 1A DM. ICAs
are present in the majority of individuals (>75%) diagnosed with new-onset
type 1A DM, in a significant minority of individuals with newly diagnosed type 2
DM, and occasionally in individuals with GDM (<5%). ICAs are present in 3 to
4% of first-degree relatives of individuals with type 1A DM. In conjunction
with impaired insulin secretion on intravenous glucose tolerance testing, they
predict a >50% risk of developing type 1A DM within 5 years. Without this
impairment in insulin secretion, the presence of ICAs predicts a 5-year risk of
<25%. Based on these data, the risk of a first-degree relative developing
type 1A DM is relatively low, and even ICA-positive individuals are not
destined to develop diabetes. At present, the ICAs are used predominantly as a
research tool and not in clinical practice, in part because of the technically
demanding nature of the assay but also because no treatments have been proven to
prevent the occurrence or progression of type 1A DM.
Environmental Factors
Numerous
environmental events have been proposed to trigger the autoimmune process in
genetically susceptible individuals; however, none have been conclusively
linked to diabetes. Identification of an environmental trigger has been
difficult because the event may precede the onset of DM by several years (Fig.
333-5). Putative environmental triggers include viruses (coxsackie and rubella
most prominently), early exposure to bovine milk proteins, and nitrosourea
compounds. Epidemiologic studies have noted an association between bovine milk
intake and type 1A DM; studies are ongoing to investigate a possible
relationship between exposure to bovine milk and the autoimmune process of type
1A DM.
Prevention of Type 1A DM
A
number of interventions have successfully delayed or prevented diabetes in
animal models. Some interventions have targeted the immune system directly
(immunosuppression, selective T cell subset deletion, induction of immunologic
tolerance to islet proteins), whereas others have prevented islet cell death by
blocking cytotoxic cytokines or increasing islet resistance to the destructive
process. Though results in animal models are promising, most of these
interventions have not been successful in preventing type 1A DM in humans.
Clinical trials of several interventions are underway in the United States and
Europe. The Diabetes Prevention Trial-type 1 is being conducted to determine
whether administering insulin to individuals at high risk for developing type
1A DM can induce immune tolerance and alter the autoimmune process of type 1A
DM.
TYPE 2 DM
Type
2 DM is a heterogeneous disorder with a complex etiology that develops in
response to genetic and environmental influences. Central to the development of
type 2 DM are insulin resistance and abnormal insulin secretion. Although
controversy remains regarding the primary defect, most studies support the view
that insulin resistance precedes insulin secretory defects.
![]() Genetics
Genetics
Type
2 DM has a strong genetic component. Although the major genes that predispose
to this disorder have yet to be identified, it is clear that the disease is
polygenic and multifactorial. Various genetic loci contribute to
susceptibility, and environmental factors (such as nutrition and physical
activity) further modulate phenotypic expression of the disease. The
concordance of type 2 DM in identical twins is between 70 and 90%. Individuals
with a parent with type 2 DM have an increased risk of diabetes; if both
parents have type 2 DM, the risk in offspring may reach 40%. Insulin
resistance, as demonstrated by reduced glucose utilization in skeletal muscle,
is present in many nondiabetic, first-degree relatives of individuals with type
2 DM. However, definition of the genetic abnormalities of type 2 DM remains a
challenge because the genetic defect in insulin secretion or action may not
manifest itself unless an environmental event or another genetic defect, such
as obesity, is superimposed.
The
identification of individuals with mutations in various molecules involved in
insulin action (e.g., the insulin receptor and enzymes involved in glucose homeostasis)
has been useful for characterizing key steps in insulin action. However,
mutations in these molecules account for a very small fraction of type 2 DM.
Likewise, genetic defects in proteins involved in insulin secretion have not
been found in most individuals with type 2 DM. Genome-wide scanning for
mutations or polymorphisms associated with type 2 DM is being used in an effort
to identify genes associated with type 2 DM.
Pathophysiology
Type
2 DM is characterized by three pathophysiologic abnormalities: impaired insulin
secretion, peripheral insulin resistance, and excessive hepatic glucose
production. Obesity, particularly visceral or central, is very common in type 2
DM. Insulin resistance associated with obesity augments the genetically determined
insulin resistance of type 2 DM. Adipocytes secrete a number of biologic
products (leptin, tumor necrosis factor ![]() ,
free fatty acids) that modulate processes such as insulin secretion, insulin
action, and body weight and may contribute to the insulin resistance. In the
early stages of the disorder, glucose tolerance remains normal, despite insulin
resistance, because the pancreatic beta cells compensate by increasing insulin
output. As insulin resistance and compensatory hyperinsulinemia progress, the
pancreatic islets become unable to sustain the hyperinsulinemic state. IGT,
marked by elevations in postprandial glucose, then develops. A further decline
in insulin secretion and an increase in hepatic glucose production lead to
overt diabetes with fasting hyperglycemia. Ultimately, beta cell failure may
ensue.
,
free fatty acids) that modulate processes such as insulin secretion, insulin
action, and body weight and may contribute to the insulin resistance. In the
early stages of the disorder, glucose tolerance remains normal, despite insulin
resistance, because the pancreatic beta cells compensate by increasing insulin
output. As insulin resistance and compensatory hyperinsulinemia progress, the
pancreatic islets become unable to sustain the hyperinsulinemic state. IGT,
marked by elevations in postprandial glucose, then develops. A further decline
in insulin secretion and an increase in hepatic glucose production lead to
overt diabetes with fasting hyperglycemia. Ultimately, beta cell failure may
ensue.
Metabolic Abnormalities
Insulin Resistance
This
is caused by the decreased ability of insulin to act effectively on peripheral
target tissues (especially muscle and liver) and is a prominent feature of type
2 DM. This resistance is relative, since supernormal levels of circulating
insulin will normalize the plasma glucose. Insulin dose-response curves exhibit
a rightward shift, indicating reduced sensitivity, and a reduced maximal
response, indicating an overall decrease in maximum glucose utilization (30 to
60% lower than normal individuals). Resistance to the action of insulin impairs
glucose utilization by insulin-sensitive tissues and increases hepatic glucose
output-both effects contributing to the hyperglycemia of diabetes. Increased
hepatic glucose output predominantly accounts for increased FPG levels, whereas
decreased peripheral glucose usage results in postprandial hyperglycemia. In
skeletal muscle, there is a greater impairment in nonoxidative glucose usage
(glycogen formation) than in oxidative glucose metabolism through glycolysis.
Glucose usage in insulin-independent tissues is not decreased in type 2 DM.
The
precise molecular mechanism of insulin resistance in type 2 DM has yet to be
elucidated. Insulin receptor levels and tyrosine kinase activity in skeletal
muscle are reduced, but these alterations are most likely secondary to
hyperinsulinemia and are not a primary defect. Therefore, postreceptor defects
are believed to play the predominant role in insulin resistance (Fig. 333-4).
Polymorphisms in IRS-1 may be associated with glucose intolerance, raising the
possibility that polymorphisms in various postreceptor molecules may combine to
create an insulin-resistant state.
A
current focus for the pathogenesis of insulin resistance focuses on a PI-3
kinase signaling defect, which causes reduced translocation of GLUT4 to the
plasma membrane, among other abnormalities. Of note, not all insulin signal
transduction pathways are resistant to the effects of insulin (e.g., those
controlling cell growth and differentiation). Consequently, hyperinsulinemia
may actually increase the insulin action through these pathways.
Another
emerging theory proposes that elevated levels of free fatty acids, a common
feature of obesity, may contribute to the pathogenesis of type 2 DM in several
different ways. Free fatty acids can impair glucose utilization in skeletal
muscle, promote glucose production by the liver, and impair beta cell function.
Impaired Insulin Secretion
Insulin
secretion and sensitivity are interrelated (Fig. 333-6). In type 2 DM, insulin
secretion initially increases in response to insulin resistance in order to
maintain normal glucose tolerance. Initially, the insulin secretory defect is
mild and selectively involves glucose-stimulated insulin secretion. The
response to other nonglucose secretagogues, such as arginine, is preserved.
Eventually, the insulin secretory defect progresses to a state of grossly
inadequate insulin secretion. Some endogenous insulin production continues, but
the amount secreted is less than the amount secreted by normal individuals at
the same plasma glucose concentration.
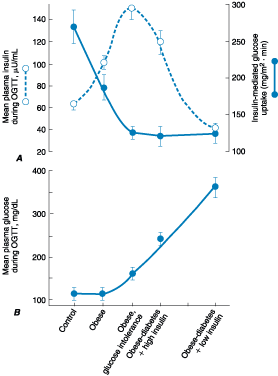
Figure 333-6: Metabolic changes during the development
of type 2 diabetes. A. The mean plasma insulin and insulin-mediated glucose
uptake during an oral glucose tolerance test (OGTT). B. The mean plasma glucose
during an OGTT. On the x-axis are groups of: control individuals, obese
individuals, obese and glucose intolerant individuals, obese individuals with
diabetes and high insulin, and obese individuals with diabetes and low
insulin.(From RA DeFronzo: Lilly lecture. The triumvirate: Beta-cell, muscle,
liver: A collusion responsible for NIDDM. Diabetes 37:667, 1998, with
permission.)
The
reason(s) for the decline in insulin secretory capacity in type 2 DM is
unclear. Despite the assumption that a second genetic defect-superimposed upon
insulin resistance-leads to beta cell failure, intense genetic investigation
has so far excluded mutations in islet candidate genes. Islet amyloid
polypeptide or amylin is cosecreted by the beta cell and likely forms the
amyloid fibrillar deposit found in the islets of individuals with longstanding
type 2 DM. Whether such islet amyloid deposits are a primary or secondary event
is not known. The metabolic environment may also impact islet function
negatively. For example, chronic hyperglycemia paradoxically impairs islet
function ("glucose toxicity") and leads to a worsening of
hyperglycemia. Improvement in glycemic control is often associated with
improved islet function. In addition, elevation of free fatty acid levels
("lipotoxicity") also worsens islet function.
Increased Hepatic Glucose Production
The
liver maintains plasma glucose during periods of fasting through glycogenolysis
and gluconeogenesis using substrates derived from skeletal muscle and fat
(alanine, lactate, glycerol, and fatty acids). Insulin promotes the storage of
glucose as hepatic glycogen and suppresses gluconeogenesis. In type 2 DM,
insulin resistance in the liver arises from the failure of hyperinsulinemia to
suppress gluconeogenesis, which results in fasting hyperglycemia and decreased
glucose storage by the liver in the postprandial state. Increased hepatic
glucose production occurs early in the course of diabetes, though likely after
the onset of insulin secretory abnormalities and insulin resistance in skeletal
muscle.
Insulin Resistance Syndromes
It
is likely that the insulin resistance condition comprises a spectrum of
disorders, with hyperglycemia representing one of the most readily diagnosed
features. Syndrome X is a term used to describe a constellation of
metabolic derangements that includes insulin resistance, hypertension,
dyslipidemia, central or visceral obesity, endothelial dysfunction, and
accelerated cardiovascular disease. Epidemiologic evidence supports
hyperinsulinemia as a marker for coronary artery disease risk, though an
etiologic role has not been demonstrated.
A
number of forms of severe insulin resistance may be associated with a phenotype
similar to that in type 2 DM or IGT (Table 333-1). Acanthosis nigricans
and signs of hyperandrogenism (hirsutism, acne, and oligomenorrhea) are common
physical features. In addition to rare genetic syndromes seen in early
childhood, two distinct syndromes of severe insulin resistance have been
described in adults: (1) type A, which affects young women and is characterized
by severe hyperinsulinemia, obesity, and features of hyperandrogenism; and (2)
type B, which affects middle-aged women and is characterized by severe
hyperinsulinemia, features of hyperandrogenism, and autoimmune disorders.
Individuals with the type A insulin resistance syndrome have an undefined
defect in the insulin signaling pathway; individuals with the type B insulin
resistance syndrome have autoantibodies directed at the insulin receptor. These
receptor autoantibodies may block insulin binding or may stimulate the insulin
receptor, leading to intermittent hypoglycemia.
Polycystic ovary syndrome (PCOS) is a common disorder that affects
premenopausal women and is characterized by chronic anovulation and
hyperandrogenism. Insulin resistance is seen in a significant subset of women
with PCOS, and the disorder substantially increases the risk for type 2 DM,
independent of the effects of obesity. Both metformin and thiazolidinediones
may attenuate hyperinsulinemia, ameliorate hyperandrogenism, and induce
ovulation, but are not approved for this indication.
Prevention
Because
type 2 DM is preceded by a period of IGT, a number of life-style modifications
and pharmacologic agents have been suggested to prevent or delay its onset.
Individuals with a strong family history or those at high risk for developing
DM should be strongly encouraged to maintain a normal body mass index and to
engage in regular physical activity. Beyond this general advice, however, there
are no specific interventions proven to prevent type 2 DM. Clinical trials of
various interventions in individuals with IGT or early DM are underway in the
United States and worldwide.
MODY: GENETICALLY DEFINED, MONOGENIC FORMS OF
DIABETES MELLITUS
Several
monogenic forms of DM have recently been identified. MODY comprises a
phenotypically and genetically heterogeneous subtype of DM. Onset of the
disease typically occurs between the ages of 10 and 25. Five different variants
of MODY, due to mutations in genes encoding islet cell transcription factors or
glucokinase (Fig. 333-3), have been identified so far, and all are transmitted
as autosomal dominant disorders (Table 333-1). MODY 2, the most common variant,
is caused by mutations in the glucokinase gene. Glucokinase catalyzes the
formation of glucose-6-phosphate from glucose, a reaction that is important for
glucose sensing by the beta cells and for glucose utilization by the liver. As
a result of glucokinase mutations, higher glucose levels are required to elicit
insulin secretory responses, thus altering the set point for insulin secretion.
MODY 1, MODY 3, and MODY 5 are caused by mutations in the hepatocyte nuclear transcription
factors HNF-4![]() ,
HNF-1
,
HNF-1![]() ,
and HNF-1
,
and HNF-1![]() ,
respectively. As their names imply, these transcription factors are expressed
in the liver but also in other tissues, including the pancreatic islets. The
mechanisms by which such mutations lead to DM is not well understood, but it is
likely that these factors affect islet development or the transcription of
genes that are important in stimulating insulin secretion. MODY 4 is a rare
variant caused by mutations in the insulin promoter factor (IPF-1), which is a
transcription factor that regulates both pancreatic development and insulin
gene transcription. Homozygous inactivating mutations lead to pancreatic
agenesis, whereas heterozygous mutations result in early-onset DM. Studies of
populations with type 2 DM suggest that mutations in the glucokinase gene and
various islet cell transcription factors do not account for ordinary type 2 DM.
Nevertheless, elucidation of the molecular genetics underlying these rare forms
of DM has been important in identifying critical steps in the control of
pancreatic beta cell function.
,
respectively. As their names imply, these transcription factors are expressed
in the liver but also in other tissues, including the pancreatic islets. The
mechanisms by which such mutations lead to DM is not well understood, but it is
likely that these factors affect islet development or the transcription of
genes that are important in stimulating insulin secretion. MODY 4 is a rare
variant caused by mutations in the insulin promoter factor (IPF-1), which is a
transcription factor that regulates both pancreatic development and insulin
gene transcription. Homozygous inactivating mutations lead to pancreatic
agenesis, whereas heterozygous mutations result in early-onset DM. Studies of
populations with type 2 DM suggest that mutations in the glucokinase gene and
various islet cell transcription factors do not account for ordinary type 2 DM.
Nevertheless, elucidation of the molecular genetics underlying these rare forms
of DM has been important in identifying critical steps in the control of
pancreatic beta cell function.
COMPLICATIONS OF DM
ACUTE COMPLICATIONS
Diabetic
ketoacidosis (DKA) and nonketotic hyperosmolar state (NKHS) are acute
complications of diabetes. DKA is seen primarily in individuals with type 1 DM,
and NKHS is seen in individuals with type 2 DM. Both disorders are associated
with absolute or relative insulin deficiency, volume depletion, and altered
mental status. DKA and NKHS exist along a continuum of hyperglycemia, with or
without ketosis. The metabolic similarities and differences in DKA and NKHS are
highlighted in Table 333-4. Both disorders are associated with potentially
serious complications if not promptly diagnosed and treated.
Table 333-4: Laboratory Values in Diabetic
Ketoacidosis (DKA) and Nonketotic Hyperosmolar
States (NKHS) (Representative Ranges at
Presentation)
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
DIABETIC KETOACIDOSIS
Clinical Features
The
symptoms and physical signs of DKA are listed in Table 333-5. DKA may be the
initial symptom complex that leads to a diagnosis of type 1 DM, but more
frequently it occurs in individuals with established diabetes. Nausea and
vomiting are often prominent, and their presence in an individual with diabetes
warrants laboratory evaluation for DKA. Abdominal pain may be severe and
sometimes suggests acute pancreatitis or ruptured viscous. Hyperglycemia leads
to glucosuria, volume depletion, tachycardia, and possibly hypotension.
Kussmaul respirations and an acetone odor on the patient's breath (both
secondary to metabolic acidosis) are classic signs of the disorder. Lethargy
and central nervous system depression may evolve into coma with severe DKA.
Cerebral edema, an extremely serious complication of DKA, is seen most
frequently in children. Signs of infection, which may precipitate DKA, should
be sought on physical examination, even in the absence of fever.
Table 333-5: Manifestations of Diabetic
Ketoacidosis
|
||||||||||||||||||||
|
|
|||||||||||||||||||
Pathophysiology
DKA
results from insulin deficiency combined with counterregulatory hormone excess
(glucagon, catecholamines, cortisol, and growth hormone). Both insulin
deficiency and glucagon excess, in particular, are necessary for DKA to
develop. The hyperglycemia of DKA results from increased hepatic glucose
production (gluconeogenesis and glycogenolysis) and impaired peripheral glucose
utilization. The decreased ratio of insulin to glucagon promotes
gluconeogenesis, glycogenolysis, and ketone body formation in the liver, as
well as increasing substrate delivery from fat and muscle (free fatty acids, amino
acids) to the liver.
The
combination of insulin deficiency and hyperglycemia reduces the hepatic level
of fructose-2,6-phosphate, which alters the activity of phosphofructokinase and
fructose-1,6-bisphosphatase. Glucagon excess decreases the activity of pyruvate
kinase, whereas insulin deficiency increases the activity of
phosphoenolpyruvate carboxykinase. These hepatic changes shift the handling of
pyruvate toward glucose synthesis and away from glycolysis. Glycogenolysis is
promoted by the increased levels of glucagon and catecholamines in the face of
low insulin levels. Insulin deficiency also reduces levels of the GLUT4 glucose
transporter, which impairs glucose uptake into skeletal muscle and fat and
reduces intracellular glucose metabolism (Fig. 333-4).
Ketosis results from a marked increase in free fatty acid release
from adipocytes, with a resulting shift toward ketone body synthesis in the
liver. Reduced insulin levels, in combination with elevations in catecholamines
and growth hormone, lead to an increase in lipolysis and release of free fatty
acids. Normally, these free fatty acids are converted to triglycerides or very
low density lipoproteins (VLDL) in the liver, but in DKA, hyperglucagonemia
alters hepatic metabolism to favor ketone body formation, through activation of
the enzyme carnitine palmitoyltransferase I. This enzyme is crucial for
regulating fatty acid transport into the mitochondria, where beta oxidation and
conversion to ketone bodies occurs. At physiologic pH, ketone bodies exist as
ketoacids, which are neutralized by bicarbonate. As bicarbonate stores are
depleted, metabolic acidosis ensues. Increased lactic acid production also
contributes to the acidosis. The increased free fatty acids result in increased
triglyceride production and increased hepatic production of VLDL. VLDL
clearance is also reduced because the activity of insulin-sensitive lipoprotein
lipase is decreased. Hypertriglyceridemia may be severe enough to cause
pancreatitis.
DKA
can be precipitated by inadequate levels of plasma insulin for a variety of
reasons (Table 333-5). Most commonly, DKA is precipitated when relatively
insufficient insulin is available when insulin requirements increase, as might
occur during a concurrent illness. Failure to augment insulin therapy
appropriately by the patient or health care team compounds the problem.
Occasionally, complete omission of insulin by the patient or health care team
(in a hospitalized patient with type 1 DM) precipitates DKA. Patients using
insulin infusion devices with short-acting insulin have a greater potential for
DKA, since even a brief interruption in insulin delivery (e.g., mechanical
malfunction) quickly leads to insulin deficiency.
Laboratory Abnormalities and Diagnosis
The
timely diagnosis of DKA is crucial and allows for prompt initiation of therapy.
DKA is characterized by hyperglycemia, ketosis, and metabolic acidosis
(increased anion gap) along with a number of secondary metabolic derangements
(Table 333-4)). Serum bicarbonate is frequently <10 mmol/L, and arterial pH
ranges between 6.8 and 7.3, depending on the severity of the acidosis. Despite
a total-body potassium deficit, the serum potassium at presentation is
typically at the high end of the normal range or mildly elevated, secondary to
the acidosis. Total-body stores of sodium, chloride, phosphorous, and magnesium
are also reduced in DKA, but are not accurately reflected by their levels in
the serum. Elevated blood urea nitrogen (BUN) and serum creatinine levels
reflect intravascular volume depletion. Interference from acetoacetate may
falsely elevate the serum creatinine measurement. Leukocytosis,
hypertriglyceridemia, and hyperlipoproteinemia are commonly found as well.
Hyperamylasemia may suggest a diagnosis of pancreatitis, especially when accompanied
by abdominal pain. However, in DKA the amylase is usually of salivary origin
and thus is not diagnostic of pancreatitis.
The
measured serum sodium is reduced as a consequence of the hyperglycemia [1.6 meq
(1.6 mmol/L) reduction in serum sodium for each 100 mg/dL (5.6 mmol/L) rise in
the serum glucose]. A normal serum sodium in the setting of DKA indicates a
more profound water deficit. In "conventional" units, the calculated
serum osmolality [2 × (serum sodium + serum potassium) + plasma glucose (mg/dL)/18
+ BUN/2.8] is mildly to moderately elevated, though to a lesser degree than
that found in NKHS hyperosmolar state (see below).
In
DKA, the ketone body, ![]() -hydroxybutyrate,
is synthesized at a threefold greater rate than acetoacetate; however, the
latter ketone body is preferentially detected by a commonly used ketosis
detection reagent (nitroprusside). Serum ketones are present at significant
levels (usually positive at serum dilution of 1:8 or greater). The
nitroprusside tablet, or stick, is often used to detect urine ketones; certain
medications such as captopril or penicillamine may cause false-positive reactions.
Serum or plasma assays for
-hydroxybutyrate,
is synthesized at a threefold greater rate than acetoacetate; however, the
latter ketone body is preferentially detected by a commonly used ketosis
detection reagent (nitroprusside). Serum ketones are present at significant
levels (usually positive at serum dilution of 1:8 or greater). The
nitroprusside tablet, or stick, is often used to detect urine ketones; certain
medications such as captopril or penicillamine may cause false-positive reactions.
Serum or plasma assays for ![]() -hydroxybutyrate
more accurately reflect the true ketone body level.
-hydroxybutyrate
more accurately reflect the true ketone body level.
The
metabolic derangements of DKA exist along a spectrum, beginning with mild
acidosis with moderate hyperglycemia evolving into more severe findings. The
degree of acidosis and hyperglycemia do not necessarily correlate closely, as a
variety of factors determine the level of hyperglycemia (oral intake, urinary
glucose loss). Ketonemia is a consistent finding in DKA and distinguishes it
from simple hyperglycemia.
![]() Treatment
Treatment
The
management of DKA is outlined in Table 333-6. After initiating intravenous
fluid replacement and insulin therapy, the agent or event that precipitated the
episode of DKA should be sought and aggressively treated. If the patient is
vomiting or has altered mental status, a nasogastric tube should be inserted to
prevent aspiration of gastric contents. Central to successful treatment of DKA
is careful patient monitoring and frequent reassessment to ensure that the
patient and the metabolic derangements are improving. A comprehensive flow
sheet should record chronologic changes in vital signs, fluid intake and
output, and laboratory values as a function of insulin administered.
Table 333-6: Management of Diabetic Ketoacidosis
|
After
the initial bolus of normal saline, replacement of the sodium and free water
deficit is carried out over the next 24 h (fluid deficit is often 3 to 5 L).
When hemodynamic stability and adequate urine output are achieved, intravenous
fluids should be switched to 0.45% saline at a rate of 200 to 300 mL/h,
depending on the calculated volume deficit. The change to 0.45% saline helps
reduce the trend toward hyperchloremia later in the course of DKA.
Alternatively, initial use of lactated Ringer's intravenous solution may reduce
the hyperchloremia that commonly occurs with normal saline.
A
bolus of intravenous or intramuscular insulin (10 to 20 units) should be
administered immediately (Table 333-6)), and subsequent treatment should
provide continuous and adequate levels of circulating insulin. Intravenous
administration is preferred, because it assures rapid distribution and allows
adjustment of the infusion rate as the patient responds to therapy. Intravenous
insulin should be continued until the acidosis resolves and the patient is
metabolically stable. As the acidosis and insulin resistance associated with
DKA resolve, the insulin infusion rate can be decreased (to 1 to 4 units/h).
Intermediate or long-acting insulin, in combination with subcutaneous regular
insulin, should be administered as soon as the patient resumes eating, as this
facilitates transition to an outpatient insulin regimen and reduces length of
hospital stay. It is crucial to continue the insulin infusion until adequate insulin
levels are achieved by the subcutaneous route. Even relatively brief periods of
inadequate insulin administration in this transition phase may allow for DKA
relapse.
Hyperglycemia
usually improves at a rate of 4.2 to 5.6 mmol/L (75 to 100 mg/dL per hour) as a
result of insulin-mediated glucose disposal, reduced hepatic glucose release,
and rehydration. The latter reduces catecholamines, increases urinary glucose
loss, and expands the intravascular volume. The decline in the plasma glucose
within the first 1 to 2 h may be more rapid and is mostly related to volume
expansion. When the plasma glucose reaches 13.9 mmol/L (250 mg/dL), glucose
should be added to the 0.45% saline infusion to maintain the plasma glucose in
the 11.1 to 13.9 mmol/L (200 to 250 mg/dL) range, and the insulin infusion
should be continued. Ketoacidosis begins to resolve as insulin reduces
lipolysis, increases peripheral ketone body use, suppresses hepatic ketone body
formation, and promotes bicarbonate regeneration. However, the acidosis and
ketosis resolve at a slower rate than does the hyperglycemia. As ketoacidosis
improves, ![]() -hydroxybutyrate
is converted to acetoacetate. Ketone body levels may appear to increase if
measured by laboratory assays that use the nitroprusside reaction, which only
detects acetoacetate and acetone levels. The improvement in acidosis and anion
gap, a result of bicarbonate regeneration and decline in ketone bodies, is
reflected by a rise in the serum bicarbonate level and the arterial pH.
Depending on the rise of serum chloride, the anion gap (but not bicarbonate)
will normalize. A hyperchloremic acidosis [serum bicarbonate of 15 to 18 mmol/L
(15 to 18 meq/L)] often follows successful treatment and is minimized by the
use of hypotonic intravenous solutions. This gradually resolves as the kidney
regenerates bicarbonate and excretes chloride.
-hydroxybutyrate
is converted to acetoacetate. Ketone body levels may appear to increase if
measured by laboratory assays that use the nitroprusside reaction, which only
detects acetoacetate and acetone levels. The improvement in acidosis and anion
gap, a result of bicarbonate regeneration and decline in ketone bodies, is
reflected by a rise in the serum bicarbonate level and the arterial pH.
Depending on the rise of serum chloride, the anion gap (but not bicarbonate)
will normalize. A hyperchloremic acidosis [serum bicarbonate of 15 to 18 mmol/L
(15 to 18 meq/L)] often follows successful treatment and is minimized by the
use of hypotonic intravenous solutions. This gradually resolves as the kidney
regenerates bicarbonate and excretes chloride.
Potassium
stores are depleted in DKA [estimated deficit 3 to 5 mmol/kg (3 to 5 meq/kg)],
but the serum potassium may be normal or even elevated at the time of
presentation. During treatment with insulin and fluids, various factors
contribute to the development of hypokalemia. These include insulin-mediated
potassium transport into cells, resolution of the acidosis (which also promotes
potassium entry into cells), and urinary loss of potassium salts of organic
acids. Thus, potassium repletion should commence as soon as adequate urine
output and a normal serum potassium are documented. If the initial serum
potassium level is elevated, then potassium repletion should be delayed until
the potassium falls into the normal range. Inclusion of 20 to 40 meq of
potassium in each liter of intravenous fluid is reasonable, but additional
potassium supplements may also be required. To reduce the amount of chloride
administered, potassium phosphate or acetate can be substituted for the
chloride salt. The goal is to maintain the serum potassium >3.5 mmol/L (3.5
meq/L).
Despite
a bicarbonate deficit, bicarbonate replacement is not usually necessary or
advisable. In fact, theoretical arguments suggest that bicarbonate
administration and rapid reversal of acidosis may impair cardiac function,
impair tissue oxygenation, and promote hypokalemia. The results of most
clinical trials do not support the routine use of bicarbonate replacement. In
the presence of severe acidosis (arterial pH < 7.0 or hypotension
unresponsive to fluid resuscitation), some physicians administer bicarbonate
[50 to 150 mmol/L (meq/L) of sodium bicarbonate in 250 mL of 0.45% saline over
1 to 2 h until the serum bicarbonate rises to approximately 10 mmol/L (meq/L)].
Hypophosphatemia may result from increased glucose usage, but randomized clinical
trials have not demonstrated that phosphate replacement is beneficial in DKA.
If the serum phosphate is < 0.32 mmol/L (1.0 mg/dl), then phosphate
supplement should be considered and the serum calcium monitored. Hypomagnesemia
may develop during DKA therapy and may also require supplementation.
With
appropriate therapy, the mortality of DKA is low (<5%) and is related more
to the underlying or precipitating event, such as infection or myocardial
infarction. The major nonmetabolic complication of DKA therapy is cerebral
edema, which most often develops in children as DKA is resolving. The etiology
and optimal therapy for cerebral edema are not well established, but
overreplacement of free water should be avoided. Venous thrombosis and adult
respiratory distress syndrome occasionally complicate DKA.
Following
successful treatment of DKA, the physician and patient should review the
sequence of events that led to DKA to prevent future recurrences. Foremost is
patient education about the symptoms of DKA, its precipitating factors, and the
management of diabetes during a concurrent illness. During illness or when oral
intake is compromised, patients should: (1) frequently measure the capillary
blood glucose; (2) measure urinary ketones when the serum glucose >16.5
mmol/L (300 mg/dL); (3) drink fluids to maintain hydration; (4) continue or
increase insulin; and (5) seek medical attention if dehydration, persistent
vomiting, or uncontrolled hyperglycemia develop. In this way, early DKA can be
detected and treated appropriately on an outpatient basis.
NONKETOTIC HYPEROSMOLAR STATE
Clinical Features
NKHS
is most commonly seen in elderly individuals with type 2 DM. Its most prominent
features include polyuria; orthostatic hypotension; and a variety of neurologic
symptoms that include altered mental status, lethargy, obtundation, seizure,
and possibly coma. The prototypical patient is a mildly diabetic, elderly
individual with a several week history of polyuria, weight loss, and diminished
oral intake that culminates in mental confusion, lethargy, or coma. The
physical examination reflects profound dehydration and hyperosmolality and
reveals hypotension, tachycardia, and altered mental status. Notably absent are
symptoms of nausea, vomiting, and abdominal pain and the Kussmaul respirations
characteristic of DKA. NKHS is often precipitated by a serious, concurrent
illness such as myocardial infarction or stroke. Sepsis, pneumonia, and other
serious infections are frequent precipitants and should be sought thoroughly.
In addition, a debilitating condition (prior stroke or dementia) or social
situation that compromises water intake may contribute to the development of
the disorder. Finally, the development of NKHS can be associated with the use
of certain medications (thiazide diuretics, glucocorticoids, phenytoin).
Pathophysiology
Insulin
deficiency and inadequate fluid intake are the underlying causes of NKHS.
Insulin deficiency increases hepatic glucose production (through glycogenolysis
and gluconeogenesis) and impairs glucose utilization in skeletal muscle (see
above discussion under DKA). Hyperglycemia induces an osmotic diuresis that
leads to profound intravascular volume depletion, which is exacerbated by
inadequate fluid replacement. The absence of ketosis in NKHS is not completely
understood. Presumably, the insulin deficiency is only relative and less severe
than in DKA. Lower levels of counterregulatory hormones and free fatty acids
have been found in NKHS than in DKA in some studies. It is also possible that
the liver is less capable of ketone body synthesis or that the insulin/glucagon
ratio does not favor ketogenesis.
Laboratory Abnormalities and Diagnosis
The
laboratory features in NKHS are summarized in Table 333-4. Most notable are the
marked hyperglycemia [plasma glucose may be >55.5 mmol/L (1000 mg/dL)],
hyperosmolality (>350 mosmol/L), and prerenal azotemia. The measured serum
sodium may be normal or slightly low despite the marked hyperglycemia. The
corrected serum sodium is usually increased [add 1.6 meq to measured sodium for
each 5.6 mmol/L (100 mg/dL) rise in the serum glucose]. In contrast to DKA,
acidosis and ketonemia are absent or mild. A small anion gap metabolic acidosis
may be present secondary to increased lactic acid. Moderate ketonuria, if present,
is secondary to starvation.
![]() Treatment
Treatment
Volume
depletion and hyperglycemia are prominent features of both NKHS and DKA.
Consequently, therapy of these disorders involves several shared elements
(Table 333-6). In both disorders, careful monitoring of the patient's fluid
status, laboratory values, and insulin infusion rate is crucial. Underlying or
precipitating problems should be aggressively sought and treated. In NKHS, the
volume depletion, free water deficit, and hyperosmolality are greater than in
DKA. The patient with NKHS is usually older, more likely to have mental status
changes, and thus more likely to have a life-threatening precipitating event
with accompanying comorbidities. Even with proper treatment, NKHS has a
substantially higher mortality than DKA (up to 50% in some clinical series).
Fluid
replacement should initially stabilize the hemodynamic status of the patient (1
to 3 L of 0.9% normal saline over the first 2 to 3 h). Because the fluid
deficit in NKHS is accumulated over a period of days to weeks, the rapidity of
reversal of the hyperosmolar state must balance the need for free water
repletion and the observation that too rapid a reversal may worsen neurologic
function. If the serum sodium is >150mmol/L (150 meq/L), 0.45% saline should
be used. After hemodynamic stability is achieved, the intravenous fluid
administration is directed at reversing the free water deficit using hypotonic
fluids (0.45% saline initially then 5% dextrose in water, D5W). The
calculated free water deficit (which averages 9 to 10 L) should be reversed
over the next 1 to 2 days (infusion rates of 200 to 300 mL/h of hypotonic
solution). Potassium repletion is usually necessary and should be dictated by
repeated measurements of the serum potassium. In patients taking diuretics, the
potassium deficit can be quite large and may be accompanied by magnesium
deficiency. Hypophosphatemia may occur during therapy and can be improved by
using KPO4 and beginning nutrition.
As
in DKA, rehydration and volume expansion lower the plasma glucose initially,
but insulin is eventually required. In NKHS, patients tend to be more sensitive
to insulin than in DKA and dose requirements are not usually as large. A
reasonable regimen for NKHS begins with an intravenous insulin bolus of 5 to 10
units followed by intravenous insulin at a constant infusion rate (3 to 7
units/h). As in DKA, glucose should be added to intravenous fluid when the
plasma glucose falls to 13.9 mmol/L (250 mg/dL), and the insulin infusion rate
should be decreased to 1 to 2 units/h. The insulin infusion should be continued
until the patient has resumed eating and can be transferred to a subcutaneous
insulin regimen. The patient should be discharged from the hospital on insulin,
though some patients can later undergo a trial of oral glucose-lowering agents.
CHRONIC COMPLICATIONS
The
chronic complications of DM affect many organ systems and are responsible for
the majority of morbidity and mortality associated with the disease. Chronic
complications can be divided into vascular and nonvascular complications (Table
333-7). The vascular complications of DM are further subdivided into
microvascular (retinopathy, neuropathy, nephropathy) and macrovascular
complications (coronary artery disease, peripheral vascular disease,
cerebrovascular disease). Nonvascular complications include problems such as
gastroparesis, sexual dysfunction, and skin changes. This division is rather
arbitrary since it is likely that multiple pathogenic processes are involved in
all forms of complications.
Table 333-7: Chronic Complications of Diabetes
Mellitus
|
The
risk of chronic complications increases as a function of the duration of
hyperglycemia; they usually become apparent in the second decade of
hyperglycemia. Since type 2 DM may have a long asymptomatic period of
hyperglycemia, many individuals with type 2 DM have complications at the time
of diagnosis.
The
microvascular complications of both type 1 and type 2 DM result from chronic
hyperglycemia. Randomized, prospective clinical trials involving large numbers
of individuals with type 1 or type 2 DM have conclusively demonstrated that a
reduction in chronic hyperglycemia prevents or reduces retinopathy, neuropathy,
and nephropathy. Other incompletely defined factors also modulate the
development of complications. For example, despite longstanding DM, some
individuals never develop nephropathy or retinopathy. Many of these patients
have glycemic control that is indistinguishable from those who develop
microvascular complications. Because of these observations, it is suspected
that a genetic susceptibility for developing particular complications exists.
However, the genetic loci responsible for these susceptibilities have not yet
been identified.
Evidence
implicating a causative role for chronic hyperglycemia in the development of
macrovascular complications is less conclusive, but some results suggest a role
for chronic hyperglycemia in the development of macrovascular disease. For
example, coronary heart disease events and mortality are two to four times
greater in patients with type 2 DM. These events correlate with fasting and postprandial
plasma glucose levels as well as with the HbA1c. Other factors (dyslipidemia
and hypertension) also play important roles in macrovascular complications.
MECHANISMS OF COMPLICATIONS
Although
chronic hyperglycemia is an important etiologic factor leading to complications
of DM, the mechanism(s) by which it leads to such diverse cellular and organ
dysfunction is unknown. Three major theories, which are not mutually exclusive,
have been proposed to explain how hyperglycemia might lead to the chronic complications
of DM (Fig. 333-7).
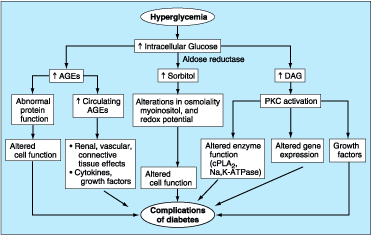
Figure 333-7: Possible molecular mechanisms of
diabetes-related complications. AGEs, advanced glycation end products; PKC,
protein kinase C; DAG, diacylglycerol; cPLA2, phospholipase A2;
Na,K-ATPase, sodium-potassium ATPase.
One
hypothesis is that increased intracellular glucose leads to the formation of
advanced glycosylation end products (AGEs) via the nonenzymatic glycosylaton of
cellular proteins. Nonenzymatic glycosylation results from the interaction of
glucose with amino groups on proteins. AGEs have been shown to cross-link
proteins (e.g., collagen, extracellular matrix proteins), accelerate atherosclerosis,
promote glomerular dysfunction, reduce nitric oxide synthesis, induce
endothelial dysfunction, and alter extracellular matrix composition and
structure. The serum level of AGEs correlates with the level of glycemia, and
these products accumulate as glomerular filtration rate declines.
A
second hypothesis proposed to explain how chronic hyperglycemia leads to
complications of DM is based on the observation that hyperglycemia increases
glucose metabolism via the sorbitol pathway. Intracellular glucose is
predominantly metabolized by phosphorylation and subsequent glycolysis, but
when intracellular glucose is increased, some glucose is converted to sorbitol
by the enzyme aldose reductase. Increased sorbitol concentrations affect
several aspects of cellular physiology (decreased myoinositol, altered redox
potential) and may lead to cellular dysfunction. However, testing of this
theory in humans, using aldose reductase inhibitors, has not demonstrated
beneficial effects on clinical endpoints of retinopathy, neuropathy, or
nephropathy.
A
third hypothesis proposes that hyperglycemia increases the formation of
diacylglycerol leading to activation of certain isoforms of protein kinase C
(PKC), which, in turn, affect a variety of cellular events that lead to
DM-related complications. For example, PKC activation by glucose alters the
transcription of genes for fibronectin, type IV collagen, contractile proteins,
and extracellular matrix proteins in endothelial cells and neurons in vitro.
Growth factors appear to play an important role in DM-related complications.
Vascular endothelial growth factor (VEGF) is increased locally in diabetic
proliferative retinopathy and decreases after laser photocoagulation.
Transforming growth factor ![]() (TGF-
(TGF-![]() )
is increased in diabetic nephropathy and appears to stimulate basement membrane
production of collagen and fibronectin by mesangial cells. Other growth
factors, such as platelet-derived growth factor, epidermal growth factor,
insulin-like growth factor I, growth hormone, basic fibroblast growth factor,
and even insulin, have been suggested to play a role in DM-related
complications.
)
is increased in diabetic nephropathy and appears to stimulate basement membrane
production of collagen and fibronectin by mesangial cells. Other growth
factors, such as platelet-derived growth factor, epidermal growth factor,
insulin-like growth factor I, growth hormone, basic fibroblast growth factor,
and even insulin, have been suggested to play a role in DM-related
complications.
Although
hyperglycemia serves as the initial trigger for complications of diabetes, it
is still unknown whether the same pathophysiologic processes are operative in
all complications or whether certain processes predominate in certain organs.
Finally, oxidative stress and free radical generation, as a consequence of the
hyperglycemia, may also promote the development of complications.
GLYCEMIC CONTROL AND COMPLICATIONS
The
Diabetes Control and Complications Trial (DCCT) provided definitive proof that
reduction in chronic hyperglycemia can prevent many of the early complications
of type 1 DM. This large multicenter clinical trial randomized over 1400
individuals with type 1 DM to either intensive or conventional diabetes
management, and then evaluated the development of retinopathy, nephropathy, and
neuropathy. Individuals in the intensive diabetes management group received multiple
administrations of insulin each day along with intense educational,
psychological, and medical support. Individuals in the conventional diabetes
management group received twice daily insulin injections and quarterly
nutritional, educational, and clinical evaluation. The goal in the former group
was normoglycemia; the goal in the latter group was prevention of symptoms of
diabetes. Individuals in the intensive diabetes management group achieved a
substantially lower HbA1c (7.2%) than individuals in the conventional diabetes
management group (HbA1c of 9.0%).
Results
from the DCCT demonstrated that improvement of glycemic control reduced
nonproliferative and proliferative retinopathy (47% reduction),
microalbuminuria (39% reduction), clinical nephropathy (54% reduction), and
neuropathy (60% reduction). Improved glycemic control also slowed the
progression of early diabetic complications. There was a nonsignificant trend
in reduction of macrovascular events. The results of the DCCT predicted that
individuals in the intensive diabetes management group would gain 7.7
additional years of sight, 5.8 additional years free from end-stage renal
disease (ESRD), and 5.6 years free from lower extremity amputations. If all
complications of DM were combined, individuals in the intensive diabetes
management group would experience 15.3 more years of life without significant
microvascular or neurologic complications of DM as compared to individuals who
received standard therapy. This translates into an additional 5.1 years of life
expectancy for individuals in the intensive diabetes management group. The
benefit of the improved glycemic control during the DCCT persisted even after
the study concluded and glycemic control worsened.
The
benefits of an improvement in glycemic control occurred over the entire range
of HbA1c values (Fig. 333-8), suggesting that at any HbA1c level, an
improvement in glycemic control is beneficial. Therefore, there is no threshold
beneath which the HbA1c can be reduced and the complications of DM prevented.
The clinical implication of this finding is that the goal of therapy is to
achieve an HbA1c level as close to normal as possible, without subjecting the
patient to excessive risk of hypoglycemia.
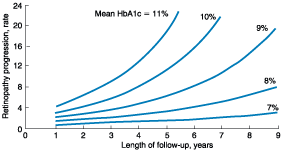
Figure 333-8: Relationship of glycemic control and
diabetes duration to diabetic retinopathy. The progression of retinopathy in
individuals in the Diabetes Control and Complications Trial is graphed as a
function of the length of follow-up with different curves for different HbA1c
values.(Adapted from The Diabetes Control and Complications Trial Research
Group, Diabetes 44:968, 1995)
Considerable
debate has emerged as to whether the DCCT findings are applicable to
individuals with type 2 DM, in whom insulin resistance, hyperinsulinemia, and
obesity predominate. Concerns have been raised that therapies associated with
weight gain and additional insulin therapy may worsen underlying insulin
resistance and hyperinsulinemia. Despite these concerns, most available data
support extrapolation of the results of the DCCT to individuals with type 2 DM.
The
United Kingdom Prospective Diabetes Study (UKPDS) studied the course of
>5000 individuals with type 2 DM for >10 years. This complex and
important study utilized multiple treatment regimens and monitored the effect
of intensive glycemic control and risk factor treatment on the development of
diabetic complications. Newly diagnosed individuals with type 2 DM were
randomized to (1) intensive management using various combinations of insulin, a
sulfonylurea, or metformin; or (2) conventional therapy using dietary
modification and pharmacotherapy with the goal of symptom prevention. In
addition, individuals were randomly assigned to different antihypertensive
regimens. Individuals in the intensive treatment arm achieved an HbA1c of 7.0%,
compared to a 7.9% HbA1c in the standard treatment group. The UKPDS
demonstrated that each percentage point reduction in HbA1c was associated with
a 35% reduction in microvascular complications, a 25% reduction in DM-related
deaths, and a 7% reduction in all-cause mortality. As in the DCCT, there was a
continuous relationship between glycemic control and development of
complications. Although there was no statistically significant effect of
glycemic control on cardiovascular complications, there was a 16% reduction in
fatal and nonfatal myocardial infarctions.
One
of the major findings of the UKPDS was the observation that strict blood
pressure control significantly reduced both macro- and microvascular
complications. In fact, the beneficial effects of blood pressure control were
greater than the beneficial effects of glycemic control. Lowering blood
pressure to moderate goals (144/82 mmHg) reduced the risk of DM-related death,
stroke, microvascular end points, retinopathy, and heart failure (risk
reductions between 32 and 56%). Improved glycemic control did not conclusively
reduce (nor worsen) cardiovascular mortality but was associated with
improvement with lipoprotein risk profiles, such as reduced triglycerides and
increased high-density lipoprotein (HDL).
Similar
reductions in the risks of retinopathy and nephropathy were also seen in a
small trial of lean Japanese individuals with type 2 DM randomized to either
intensive glycemic control or standard therapy with insulin (Kumamoto study).
These results demonstrate the effectiveness of improved glycemic control in
individuals of different ethnicity with a presumably different etiology of DM
(i.e., phenotypically different from those in the DCCT and UKPDS).
The
findings of the DCCT, UKPDS, and Kumamoto study support the idea that chronic
hyperglycemia plays a causative role in the pathogenesis of diabetic
microvascular complications. These landmark studies prove the value of
metabolic control and emphasize the importance of (1) intensive glycemic
control in all forms of DM, and (2) early diagnosis and strict blood pressure
control in type 2 DM.
OPHTHALMOLOGIC COMPLICATIONS OF DIABETES
MELLITUS
DM
is the leading cause of blindness between the ages of 20 and 74 in the United
States. The gravity of this problem is highlighted by the finding that
individuals with DM are 25 times more likely to become legally blind than individuals
without DM. Blindness is primarily the result of progressive diabetic
retinopathy and clinically significant macular edema. Diabetic retinopathy is
classified into two stages: nonproliferative and proliferative. Nonproliferative
diabetic retinopathy usually appears late in the first decade or early in
the second decade of the disease and is marked by retinal vascular
microaneurysms, blot hemorrhages, and cotton wool spots (see Plate IV-15). Mild
nonproliferative retinopathy progresses to more extensive disease,
characterized by changes in venous vessel caliber, intraretinal microvascular
abnormalities, and more numerous microaneurysms and hemorrhages. The
pathophysiologic mechanisms invoked in nonproliferative retinopathy include
loss of retinal pericytes, increased retinal vascular permeability, alterations
in retinal blood flow, and abnormal retinal microvasculature, all of which lead
to retinal ischemia.
The
appearance of neovascularization in response to retinal hypoxia is the hallmark
of proliferative diabetic retinopathy. These newly formed vessels may
appear at the optic nerve and/or macula and rupture easily, leading to vitreous
hemorrhage, fibrosis, and ultimately retinal detachment. Not all individuals
with nonproliferative retinopathy develop proliferative retinopathy, but the
more severe the nonproliferative disease, the greater the chance of evolution
to proliferative retinopathy within 5 years. This creates a clear opportunity
for early detection and treatment of diabetic retinopathy (discussed below). In
contrast, clinically significant macular edema may appear when only
nonproliferative retinopathy is present. Fluorescein angiography is often
useful to detect macular edema, which is associated with a 25% chance of
moderate visual loss over the next 3 years.
Duration
of DM and degree of glycemic control are the best predictors of the development
of retinopathy. Nonproliferative retinopathy is found in almost all individuals
who have had DM for >20 years (25% incidence with 5 years, and 80% incidence
with 15 years of type 1 DM). Although there is genetic susceptibility for
retinopathy, it confers less influence on the development of retinopathy than
either the duration of DM or the degree of glycemic control.
![]() Treatment
Treatment
The
most effective therapy for diabetic retinopathy is prevention. Intensive
glycemic control will greatly delay the development or slow the progression of
retinopathy in individuals with either type 1 or type 2 DM. Paradoxically,
during the first 6 to 12 months of improved glycemic control, established
diabetic retinopathy may transiently worsen. Fortunately, this progression is
temporary, and in the long term, improved glycemic control is associated with
less diabetic retinopathy. Individuals with known retinopathy should be
considered candidates for prophylactic photocoagulation when initiating
intensive therapy. Once advanced retinopathy is present, improved glycemic
control imparts less benefit, though adequate ophthalmologic care can prevent
most blindness.
Equally
as important as glycemic control are regular, comprehensive eye examinations
for all individuals with DM. Most diabetic eye disease can be successfully
treated if detected early. Routine, nondilated eye examinations by the primary
care provider or diabetes specialist are inadequate to detect diabetic
eye disease properly. The treatment of diabetic eye disease requires an
ophthalmologist experienced in these disorders. Laser photocoagulation is very
successful in preserving vision. Proliferative retinopathy is usually treated
with panretinal laser photocoagulation, whereas macular edema is treated with
focal laser photocoagulation. Although exercise has not been conclusively shown
to worsen proliferative diabetic retinopathy, most ophthalmologists advise
individuals with advanced diabetic eye disease to limit physical activities
associated with repeated Valsalva maneuvers. Aspirin therapy (650 mg/d) does
not appear to influence the natural history of diabetic retinopathy, but
studies of other antiplatelet agents are under way.
RENAL COMPLICATIONS OF DIABETES MELLITUS
Diabetic
nephropathy is the leading cause of ESRD in the United States and a leading cause
of DM-related morbidity and mortality. Proteinuria in individuals with DM is
associated with markedly reduced survival and increased risk of cardiovascular
disease. Individuals with diabetic nephropathy almost always have diabetic
retinopathy also.
Like
other microvascular complications, the pathogenesis of diabetic nephropathy is
related to chronic hyperglycemia (Fig. 334-7). The mechanisms by which chronic
hyperglycemia leads to ESRD, though incompletely defined, involve the
following: interaction of soluble factors (growth factors, angiotensin II,
endothelin, AGEs), hemodynamic alterations in the renal microcirculation
(glomerular hyperfiltration, increased glomerular capillary pressure), and
structural changes in the glomerulus (increased extracellular matrix, basement
membrane thickening, mesangial expansion, fibrosis). Some of these effects may
be mediated through angiotensin receptors. Smoking accelerates the decline in
renal function.
The
natural history of diabetic nephropathy is shown schematically in Fig. 333-9
and is characterized by a fairly predictable pattern of events. Although this
sequence of events was defined for individuals with type 1 DM, a similar
pattern is also likely in type 2 DM. Glomerular hyperfusion and renal
hypertrophy occur in the first years after the onset of DM and are reflected by
an increased glomerular filtration rate (GFR). During the first 5 years of DM,
thickening of the glomerular basement membrane, glomerular hypertrophy, and
mesangial volume expansion occur as the GFR returns to normal. After 5 to 10
years of type 1 DM, ~40% of individuals begin to excrete small amounts of
albumin in the urine (microalbuminuria). Microalbuminuria is defined as
30 to 300 mg/d in a 24-h collection or 30 to 300 ![]() g/mg
creatinine in a spot collection. The appearance of microalbuminuria (incipient
nephropathy) in type 1 DM is a very important predictor of progression to overt
proteinuria (>300 mg/d). Blood pressure may rise slightly at this point but
usually remains in the normal range. Once overt proteinuria is present, there
is a steady decline in GFR, and ~50% of individuals reach ESRD in 7 to 10 years.
The early pathologic changes and albumin excretion abnormalities are reversible
with normalization of plasma glucose. However, once nephropathy becomes overt,
the pathologic changes are likely irreversible.
g/mg
creatinine in a spot collection. The appearance of microalbuminuria (incipient
nephropathy) in type 1 DM is a very important predictor of progression to overt
proteinuria (>300 mg/d). Blood pressure may rise slightly at this point but
usually remains in the normal range. Once overt proteinuria is present, there
is a steady decline in GFR, and ~50% of individuals reach ESRD in 7 to 10 years.
The early pathologic changes and albumin excretion abnormalities are reversible
with normalization of plasma glucose. However, once nephropathy becomes overt,
the pathologic changes are likely irreversible.
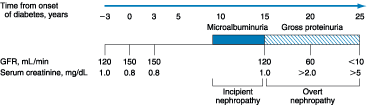
Figure 333-9: Time course of development of diabetic
nephropathy. The relationship of time from onset of diabetes, the glomerular
filtration rate (GFR), and the serum creatinine are shown.(Adapted from
DeFronzo RA, in Therapy for Diabetes Mellitus and Related Disorders, 1998)
The
nephropathy that develops in type 2 DM differs from that of type 1 DM in the
following respects: (1) microalbuminuria or overt nephropathy may be present
when type 2 DM is diagnosed, reflecting its long asymptomatic period; (2)
hypertension more commonly accompanies microalbuminuria or overt nephropathy in
type 2 DM; and (3) microalbuminuria may be less predictive of progression to
overt nephropathy in type 2 DM. Finally, it should be noted that albuminuria in
type 2 DM may be secondary to factors unrelated to DM, such as hypertension,
congestive heart failure, prostate disease, or infection.
Other
renal problems may also occur in individuals with DM. Type IV renal tubular
acidosis (hyporeninemic hypoaldosteronism) occurs in many individuals with DM
(Fig. 333-15). These individuals develop a propensity to hyperkalemia, which
may be exacerbated by medications [especially angiotensin-converting enzyme (ACE)
inhibitors]. Patients with DM are predisposed to radiocontrast-induced
nephrotoxicity. Individuals with DM undergoing radiographic procedures with
contrast dye should be well hydrated before and after dye exposure, and the
serum creatinine should be monitored for several days following the procedure.
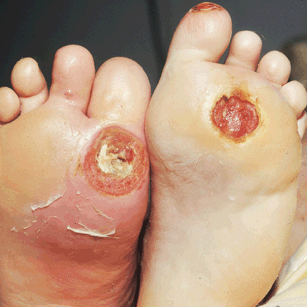
Figure 333-15: Diabetic, neuropathic
ulcers on the soles
Two large ulcers overlying the first right and second left metacarpophalangeal
joints. The patient, a 56-year-old male with diabetes mellitus of 20 years'
duration, has significant sensory neuropathy of the feet and lower legs as well
as peripheral vascular disease
![]() Treatment
Treatment
The
optimal therapy for diabetic nephropathy is prevention. As part of
comprehensive diabetes care, microalbuminuria should be detected at an early
stage when effective therapies can be instituted. The recommended strategy for
detecting microalbuminuria is outlined in Fig. 333-10. Interventions effective
in slowing progression from microalbuminuria to overt nephropathy include: (1)
near normalization of glycemia, (2) strict blood pressure control, and (3)
administration of ACE inhibitors.
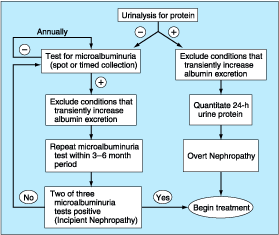
Figure 333-10: Screening for microalbuminuria.(Adapted
from DeFronzo RA, in Therapy for Diabetes Mellitus and Related Disorders, 1998)
Improved
glycemic control reduces the rate at which microalbuminuria appears and
progresses in both type 1 and type 2 DM. However, once overt nephropathy exists,
it is unclear whether improved glycemic control will slow progression of renal
disease. During the phase of declining renal function, insulin requirements may
fall as the kidney is a site of insulin degradation. Furthermore,
glucose-lowering medications (sulfonylureas and metformin) may accumulate and
are contraindicated in renal insufficiency.
Many
individuals with type 1 or type 2 DM develop hypertension. Numerous studies in
both type 1 and type 2 DM demonstrate the effectiveness of strict blood pressure
control in reducing albumin excretion and slowing the decline in renal
function. Blood pressure should be maintained at <130/85 mmHg in diabetic
individuals without proteinuria. A slightly lower blood pressure (120/80)
should be targeted for individuals with microalbuminuria or overt nephropathy.
Treatment of hypertension is discussed below.
ACE
inhibitors reduce the progression of overt nephropathy in individuals with type
1 or type 2 DM and should be prescribed in individuals with type 1 or type 2 DM
and microalbuminuria. After 2 to 3 months of therapy, measurement of
proteinuria should be repeated and the drug dose increased until either the
albuminuria disappears or the maximum dose is reached. If an ACE inhibitor has
an unacceptable side-effect profile (hyperkalemia, cough, and renal
insufficiency), angiotensin II receptor blockers and calcium channel blockers
(phenylalkylamine class) are alternatives. However, their efficacy in slowing
the fall in glomerular filtration rate is not proven. Blood pressure control
with any agent is extremely important, but a drug-specific benefit in diabetic
nephropathy, independent of blood pressure control, has been shown only for ACE
inhibitors.
A
consensus panel of the American Diabetes Association (ADA) suggests modest
restriction of protein intake in diabetic individuals with microalbuminuria
(0.8 g/kg per day, which is the adult Recommended Daily Allowance, and about
10% of the daily caloric intake). Protein intake should be restricted further
in individuals with overt diabetic nephropathy (0.6 g/kg per day), though
conclusive proof of the efficacy of protein restriction is lacking.
Nephrology
consultation should be considered after the diagnosis of early nephropathy.
Once overt nephropathy ensues, the likelihood of ESRD is very high. As compared
to nondiabetic individuals, hemodialysis in patients with DM is associated with
more frequent complications, such as hypotension (autonomic neuropathy, loss of
reflex tachycardia), more difficult vascular access, and accelerated
progression of retinopathy. Survival after the onset of ESRD is shorter in the
diabetic population compared to nondiabetics with similar clinical features.
Atherosclerosis is the leading cause of death in diabetic individuals on
dialysis, and hyperlipidemia should be aggressively treated. Renal
transplantation from a living-related donor is the preferred therapy but
requires chronic immunosuppression. Combined pancreas-kidney transplant offers
the promise of normoglycemia but requires substantial expertise.
NEUROPATHY AND DIABETES MELLITUS
Diabetic
neuropathy occurs in approximately 50% of individuals with long-standing type 1
and type 2 DM. It may manifest as polyneuropathy, mononeuropathy, and/or
autonomic neuropathy. As with other complications of DM, the development of
neuropathy correlates with the duration of diabetes and glycemic control.
Because the clinical features of diabetic neuropathy are similar to those of
other neuropathies, the diagnosis of diabetic neuropathy should be made
only after other possible etiologies are excluded (Chap. 378).
Polyneuropathy/Mononeuropathy
The
most common form of diabetic neuropathy is distal symmetric polyneuropathy.
It most frequently presents with distal sensory loss. Hyperesthesia,
parathesia, and pain also occur. Any combination of these symptoms may develop
as neuropathy progresses. Physical examination reveals sensory loss, loss of
ankle reflexes, and abnormal position sense. Parethesia is characteristically
perceived as a sensation of numbness, tingling, sharpness, or burning that
begins in the feet and spreads proximally. Neuropathic pain develops in some of
these individuals, occasionally preceded by improvement in their glycemic
control. Pain typically involves the lower extremities, is usually present at
rest, and worsens at night. Both an acute (lasting <12 months) and a chronic
form of painful diabetic neuropathy have been described. As diabetic neuropathy
progresses, the pain subsides and eventually disappears, and a sensory deficit
in the lower extremities persists.
Diabetic polyradiculopathy is a syndrome characterized by severe
disabling pain in the distribution of one or more nerve roots. It may be
accompanied by motor weakness. Intercostal or truncal radiculopathy causes pain
over the thorax or abdomen. Involvement of the lumbar plexus or femoral nerve
may cause pain in the thigh or hip and may be associated with muscle weakness
in the hip flexors or extensors (diabetic amyotrophy). Fortunately, diabetic
polyradiculopathies are usually self-limited and resolve over 6 to 12 months.
Mononeuropathy (dysfunction of isolated cranial or peripheral
nerves) is less common than polyneuropathy in DM and presents with pain and
motor weakness in the distribution of a single nerve. A vascular etiology is
favored, but the pathogenesis is unknown. Involvement of the third cranial
nerve is most common and is heralded by diplopia. Physical examination reveals
ptosis and opthalmoplegia with normal papillary constriction to light.
Sometimes cranial nerves IV, VI, or VII (Bell's palsy) are affected. Peripheral
mononeuropathies or simultaneous involvement of more than one nerve
(mononeuropathy multiplex) may also occur.
Autonomic Neuropathy
Individuals
with long-standing type 1 or 2 DM may develop signs of autonomic dysfunction
involving the cholinergic, noradrenergic, and peptidergic (peptides such as
pancreatic polypeptide, substance P, etc.) systems. DM-related autonomic
neuropathy can involve multiple systems, including: the cardiovascular,
gastrointestinal, genitourinary, sudomotor, and metabolic systems. Autonomic
neuropathies affecting the cardiovascular system cause a resting tachycardia
and orthostatic hypotension. Reports of sudden death have also been attributed
to autonomic neuropathy. Gastroparesis and bladder-emptying abnormalities are
also likely related to the autonomic neuropathy seen in DM (discussed below).
Hyperhidrosis of the upper extremities and anhidrosis of the lower extremities
result from sympathetic nervous system dysfunction. Anhidrosis of the feet can
promote dry skin with cracking, which increases the risk of skin ulceration.
Autonomic neuropathy may reduce counterregulatory hormone release, leading to
an inability to sense hypoglycemia appropriately (hypoglycemia unawareness;
Chap. 334), thereby subjecting the patient to the risk of severe hypoglycemia
and complicating efforts to improve glycemic control.
![]() Treatment
Treatment
Treatment
of diabetic neuropathy is less than satisfactory. Improved glycemic control
should be pursued and will improve nerve conduction velocity, but the symptoms
of diabetic neuropathy may not necessarily improve. Efforts to improve glycemic
control may be confounded by autonomic neuropathy and hypoglycemia unawareness.
Avoidance of neurotoxins (alcohol), supplementation with vitamins for possible
deficiencies (B12, B6, folate; Chap. 75), and symptomatic
treatment are the mainstays of therapy. Aldose reductase inhibitors do not
currently offer significant symptomatic relief. Loss of sensation in the foot
places the patient at risk for ulceration and its sequelae; consequently,
prevention of such problems is of paramount importance. Since the pain of acute
diabetic neuropathy may resolve over the first year, analgesics may be
discontinued as progressive neuronal damage from DM occurs. Chronic, painful
diabetic neuropathy is difficult to treat but may respond to tricyclic
antidepressants (amitriptyline, desipramine, nortriptyline), gabapentin,
nonsteroidal anti-inflammatory agents (avoid in renal dysfunction), and other
agents (mexilitine, phenytoin, carbamazepine, capsaicin cream). Referral to a
pain management center may be necessary.
Therapy
of orthostatic hypotension secondary to autonomic neuropathy is difficult. A
variety of agents have limited success (fludrocortisone, midodrine, clonidine,
octreotide, and yohimbine) but have significant side effects. Nonpharmacologic
maneuvers (adequate salt intake, avoidance of dehydration and diuretics, and
lower extremity support hose) may offer some benefit.
GASTROINTESTINAL/GENITOURINARY DYSFUNCTION
Long-standing
type 1 and 2 DM may affect the motility and function of gastrointestinal (GI)
and genitourinary systems. The most prominent GI symptoms are delayed gastric
emptying (gastroparesis) and altered small- and large-bowel motility
(constipation or diarrhea). Gastroparesis may present with symptoms of
anorexia, nausea, vomiting, early satiety, and abdominal bloating. Nuclear
medicine scintigraphy after ingestion of a radiolabeled meal is the best study
to document delayed gastric emptying, but noninvasive "breath tests"
following ingestion of a radiolabeled meal are under development. Though
parasympathetic dysfunction secondary to chronic hyperglycemia is important in
the development of gastroparesis, hyperglycemia itself also impairs gastric
emptying. Nocturnal diarrhea, alternating with constipation, is a common
feature of DM-related GI autonomic neuropathy. In type 1 DM, these symptoms
should also prompt evaluation for celiac sprue because of its increased
frequency. Esophageal dysfunction in long-standing DM is common but usually
asymptomatic.
Diabetic
autonomic neuropathy may lead to genitourinary dysfunction including
cystopathy, erectile dysfunction, and female sexual dysfunction (reduced sexual
desire, dyspareunia, reduced vaginal lubrication). Symptoms of diabetic
cystopathy begin with an inability to sense a full bladder and a failure to
void completely (Chap. 48). As bladder contractility worsens, bladder capacity
and the postvoid residual increase, leading to symptoms of urinary hesitancy,
decreased voiding frequency, incontinence, and recurrent urinary tract
infections. Diagnostic evaluation includes cystometry and urodynamic studies.
Erectile
dysfunction and retrograde ejaculation are very common in DM and may be one of
the earliest signs of diabetic neuropathy. Erectile dysfunction, which
increases in frequency with the age of the patient and the duration of
diabetes, may occur in the absence of other signs of diabetic autonomic
neuropathy.
![]() Treatment
Treatment
Current
treatments for these complications of DM are inadequate. Improved glycemic
control should be a primary goal, as some aspects (neuropathy, gastric
function) may improve as near-normoglycemia is achieved. Smaller, more frequent
meals that are easier to digest (liquid) and low in fat and fiber may minimize
symptoms of gastroparesis. Cisapride (10 to 20 mg before each meal) is probably
the most effective medication but has been removed from use in the U.S. market
except under special circumstances. Other agents with some efficacy include dopamine
agonists (metoclopramide, 5 to 10 mg, and domperidone, 10 to 20 mg, before each
meal) and bethanechol (10 to 20 mg before each meal). Erythromycin interacts
with the motilin receptor and may promote gastric emptying. Diabetic diarrhea
in the absence of bacterial overgrowth is treated symptomatically with
loperamide but may respond to clonidine at higher doses (0.6 mg tid) or
octreotide (50 to 75 ![]() g
tid subcutaneously). Treatment of bacterial overgrowth with antibiotics is
sometimes useful (Chap. 286).
g
tid subcutaneously). Treatment of bacterial overgrowth with antibiotics is
sometimes useful (Chap. 286).
Diabetic
cystopathy should be treated with timed voiding or self-catherization.
Medications (bethanechol) are inconsistently effective. The drug of choice for
erectile dysfunction is sildenafil, but the efficacy in individuals with DM is
slightly lower than in the nondiabetic population (Chap. 51). Sexual
dysfunction in women may be improved with use of vaginal lubricants, treatment
of vaginal infections, and systemic or local estrogen replacement.
CARDIOVASCULAR MORBIDITY AND MORTALITY
Cardiovascular
disease is increased in individuals with type 1 or type 2 DM. The Framingham
Heart Study revealed a marked increase in several cardiovascular diseases in DM
including peripheral vascular disease, congestive heart failure, coronary
artery disease, myocardial infarction, and sudden death (risk increase from
one- to fivefold). The American Heart Association recently designated DM as a
major risk factor for cardiovascular disease (same category as smoking,
hypertension, and hyperlipidemia). Because of the extremely high frequency of
underlying cardiovascular disease in individuals with diabetes (especially in
type 2 DM), evidence of atherosclerotic vascular disease should be sought in
the individual with diabetes who has symptoms suggestive of cardiac ischemia,
peripheral or carotid arterial disease, a resting electrocardiogram indicative
of prior infarction, plans to initiate an exercise program, proteinuria, or two
other cardiac risk factors (ADA recommendations). The absence of chest pain
("silent ischemia") is common in individuals with diabetes, and a
thorough cardiac evaluation is indicated in individuals undergoing major surgical
procedures.
The
increase in morbidity and mortality appears to relate to the synergism of
hyperglycemia with other cardiovascular risk factors. For example, after
controlling for all known cardiovascular risk factors, type 2 DM increases the
cardiovascular death rate by twofold in men and fourfold in women. Risk factors
for macrovascular disease in diabetic individuals include dyslipidemia,
hypertension, obesity, reduced physical activity, and cigarette smoking.
Additional risk factors specific to the diabetic population include
microalbuminuria, gross proteinuria, an elevation in serum creatinine, and
altered platelet function. Insulin resistance, as reflected by elevated serum
insulin levels, is associated with an increased risk of cardiovascular complications
in individuals with and without DM. Individuals with insulin resistance and
type 2 DM have elevated levels of plasminogen activator inhibitors (especially
PAI-1) and fibrinogen, which enhances the coagulation process and impairs
fibrinolysis, thus favoring the development of thrombosis.
Despite
proof that improved glycemic control reduces microvascular complications in DM,
it is possible that macrovascular complications may be unaffected or even
worsened by such therapy. Concerns about the anabolic and atherogenic potential
of insulin remain, since in nondiabetic individuals, higher serum insulin
levels (indicative of insulin resistance) are associated with a greater risk of
cardiovascular morbidity and mortality. In the DCCT, the number of cardiovascular
events did not differ between the standard and intensively treated groups.
However, the duration of DM in these individuals was relatively short, and the
total number of events was very low. An improvement in the lipid profile of
individuals in the intensive group [lower total and low-density lipoprotein
(LDL) cholesterol, lower triglycerides] suggested that intensive therapy may
reduce the risk of cardiovascular morbidity and mortality associated with DM.
In the UKPDS, improved glycemic control did not conclusively reduce
cardiovascular mortality. Importantly, treatment with insulin and the
sulfonylureas did not appear to increase the risk of cardiovascular disease in
individuals with type 2 DM, refuting prior claims about the atherogenic potential
of these agents.
In
addition to coronary artery disease, cerebrovascular disease is increased in
individuals with DM (threefold increase in stroke). Individuals with DM have an
increased incidence of congestive heart failure (diabetic cardiomyopathy). The
etiology of this abnormality is probably multifactorial and includes factors
such as myocardial ischemia from atherosclerosis, hypertension, and myocardial
cell dysfunction secondary to chronic hyperglycemia.
![]() Treatment
Treatment
In
general, the treatment of coronary disease is no different in the diabetic
individual (Chap. 244), though overall prognosis after myocardial infarction is
worse in the diabetic population. Revascularization procedures for coronary
artery disease, including percutaneous transluminal coronary angioplasty (PTCA)
and coronary artery bypass grafting (CABG), are less efficacious in the
diabetic individual. Initial success rates of PTCA in diabetic individuals are
similar to those in the nondiabetic population, but diabetic patients have
higher rates of restenosis and lower long-term patency and survival rates.
Perioperative mortality from CABG is not altered in DM, but both short- and
long-term survival are reduced. Recent trials indicate that diabetic
individuals with multivessel coronary artery disease or who recently suffered a
Q-wave myocardial infarction have better long-term survival with CABG than
PTCA.
Results
of studies investigating the effect of intensive diabetes management on
survival rates and cardiovascular events after myocardial infarction have been
conflicting. In the face of conflicting data, the ADA has emphasized the
importance of glycemic control and aggressive cardiovascular risk modification
in all individuals with DM. Despite past trepidation about using beta blockers
in individuals who have diabetes, these agents clearly benefit diabetic
patients after myocardial infarction, analogous to the benefit in nondiabetic
individuals. ACE inhibitors may also be particularly beneficial in reducing
mortality after myocardial infarction in patients with DM.
Antiplatelet
therapy reduces cardiovascular events in individuals with DM who have coronary
artery disease. Current recommendations by the ADA suggest the use of aspirin
as secondary prevention of additional coronary events. Although data
demonstrating efficacy in primary prevention of coronary events are lacking,
antiplatelet therapy should be considered, especially in diabetic individuals
with other coronary risk factors such as hypertension, smoking, or
hyperlipidemia. The aspirin dose (81 to 325 mg) is the same as that in
nondiabetic individuals. Aspirin therapy does not have detrimental effects on
renal function or hypertension, nor does it influence the course of diabetic
retinopathy or maculopathy.
Cardiovascular Risk Factors
Dyslipidemia
Individuals
with DM may have several forms of dyslipidemia (Chap. 344). Because of the
additive cardiovascular risk of hyperglycemia and hyperlipidemia, lipid
abnormalities should be aggressively detected and treated as part of
comprehensive diabetes care (Fig. 333-11). The most common pattern of
dyslipidemia is hypertriglyceridemia and reduced HDL cholesterol levels. DM itself
does not increase levels of LDL, but the small dense LDL particles found in
type 2 DM are more atherogenic because they are more easily glycated and
susceptible to oxidation.
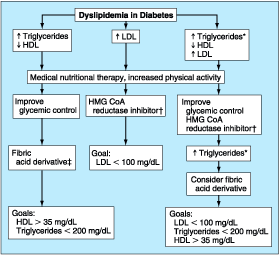
Figure 333-11: Dyslipidemia management
in diabetes. *Triglycerides increased but <400 mg/dL. ![]() Second
line treatment: fibric acid derivative or bile acid-binding resin.
Second
line treatment: fibric acid derivative or bile acid-binding resin. ![]() Alternative
treatment: high dose HMG CoA reductase inhibitor. The level of HDL in women
should be 10 mg/dl higher.
Alternative
treatment: high dose HMG CoA reductase inhibitor. The level of HDL in women
should be 10 mg/dl higher.
According
to guidelines of the ADA and the American Heart Association, the lipid profile
in diabetic individuals without cardiovascular disease (primary prevention)
should be: LDL < 3.4 mmol/L (130 mg/dL); HDL > 0.9 mmol/L (35 mg/dL) in
men and >1.2 mmol/L (45 mg/dL) in women; and triglycerides < 2.3 mmol/L
(200 mg/dL). In diabetic individuals with cardiovascular disease, the LDL goal
is < 2.6 mmol/L (100 mg/dL). Because of the risk of cardiovascular disease
in diabetic individuals, many authorities recommend that optimal lipid levels
for all individuals with DM (with or without cardiovascular disease) should be:
LDL < 2.6 mmol/L (100 mg/dL), HDL > 1.15 mmol/L (45 mg/dL) in men and
> 1.41 mmol/L (55 mg/dL) in women; and triglycerides < 2.3 mmol/L (200
mg/dL). The ADA recommends dietary modification in diabetic individuals without
cardiovascular disease and a LDL cholesterol of 2.6 to 3.3 mmol/L (100 to 129
mg/dL). If multiple cardiovascular risk factors are present, the goal should be
a LDL < 2.6 mmol/L (100 mg/dL) even without known cardiovascular disease.
Almost
all studies of diabetic dyslipidemia have been performed in individuals with
type 2 DM because of the greater frequency of dyslipidemia in this form of
diabetes. Interventional studies have shown that the beneficial effects of LDL
reduction are similar in the diabetic and nondiabetic populations. Large
prospective trials of primary and secondary intervention for coronary heart
disease have included a small number of individuals with type 2 DM, and subset
analyses have consistently found that reductions in LDL reduce cardiovascular
events and morbidity in individuals with DM. Most clinical trials used HMG CoA
reductase inhibitors, although a fibric acid derivative was also beneficial in
one trial. No prospective studies have addressed similar questions in
individuals with type 1 DM.
Based
on the guidelines provided by the ADA and the American Heart Association, the
order of priorities in the treatment of hyperlipidemia is: (1) lower the LDL
cholesterol, (2) raise the HDL cholesterol, and (3) decrease the triglycerides.
A treatment strategy depends on the pattern of lipoprotein abnormalities (Fig.
333-11). Initial therapy for all forms of dyslipidemia should include dietary
changes, as well as the same life-style modifications recommended in the
nondiabetic population (smoking cessation, control of blood pressure, weight
loss, increased physical activity). The dietary recommendations for individuals
with DM are similar to those advocated by the National Cholesterol Education
Program (Chap. 344) and include an increase in monounsaturated fat and
carbohydrates and a reduction in saturated fats and cholesterol. Though viewed
as important, the response to dietary alterations is often modest
[<0.6-mmol/L (<25-mg/dL) reduction in the LDL]. Improvement in glycemic
control will lower triglycerides and have a modest beneficial effect on raising
HDL. Most medications that improve glycemic control are useful in lowering
triglycerides and may raise the HDL slightly. Though fibric acid derivatives
have some efficacy and are well tolerated, nicotinic acid may worsen glycemic
control and increase insulin resistance; thus, niacin is relatively
contraindicated in diabetic patients on oral glucose-lowering agents. As noted
above, HMG CoA reductase inhibitors have proven benefit in patients with DM,
even with modest elevations in LDL. Combination therapy with an HMG CoA
reductase inhibitor and fibric acid derivative may be useful but increases the
possibility of myositis. Bile acid-binding resins should not be used if
hypertriglyceridemia is present.
Hypertension
Hypertension
can accelerate other complications of DM, particularly cardiovascular disease
and nephropathy. Hypertension therapy should first emphasize life-style
modifications such as weight loss, exercise, stress management, and sodium
restriction (Chap. 35). Antihypertensive agents should be selected based on the
advantages and disadvantages of the therapeutic agent in the context of an
individual patient's risk factor profile. ACE inhibitors are glucose- and
lipid-neutral and thus positively impact the cardiovascular risk profile. For
example, captopril actually improves insulin resistance, reduces LDL slightly,
and increases HDL slightly. In one study of nondiabetic individuals, the ACE
inhibitor ramipril reduced the risk of developing type 2 DM. Other effective
agents include ![]() -adrenergic
blockers (prazocin, terazosin, doxazosin), calcium channel blockers, beta
blockers (both
-adrenergic
blockers (prazocin, terazosin, doxazosin), calcium channel blockers, beta
blockers (both ![]() 1
selective and nonselective), thiazide diuretics (hydrochlorothiazide and its
derivatives), central adrenergic antagonists (clonidine, methyldopa), and
vasodilators (minoxidil, hydralazine). DM-related considerations include the
following:
1
selective and nonselective), thiazide diuretics (hydrochlorothiazide and its
derivatives), central adrenergic antagonists (clonidine, methyldopa), and
vasodilators (minoxidil, hydralazine). DM-related considerations include the
following:
1. ![]() -Adrenergic
blockers slightly improve insulin resistance and positively impact the lipid
profile, whereas beta blockers and thiazide diuretics can increase insulin
resistance, negatively impact the lipid profile, and slightly increase the risk
of developing type 2 diabetes.
-Adrenergic
blockers slightly improve insulin resistance and positively impact the lipid
profile, whereas beta blockers and thiazide diuretics can increase insulin
resistance, negatively impact the lipid profile, and slightly increase the risk
of developing type 2 diabetes.
2. Beta blockers, often
questioned because of the potential masking of hypoglycemic symptoms, are
effective agents and hypoglycemic events are rare when cardioselective (![]() 1)
agents are used.
1)
agents are used.
3. Central adrenergic
antagonists and vasodilators are lipid- and glucose-neutral.
4. Sympathetic inhibitors
and ![]() -adrenergic
blockers may be associated with orthostatic hypotension in the diabetic
individual with autonomic neuropathy.
-adrenergic
blockers may be associated with orthostatic hypotension in the diabetic
individual with autonomic neuropathy.
5. Calcium channel blockers
are glucose- and lipid-neutral, and some evidence suggests that they reduce
cardiovascular morbidity and mortality in type 2 DM, particularly in elderly
patients with systolic hypertension.
If
microalbuminuria or overt albuminuria is present, the optimal antihypertensive
agent is an ACE inhibitor. If albumin excretion is normal, then an ACE inhibitor
or other antihypertensive agent may be used. Low-dose diuretics and beta
blockers are sometimes preferred as initial agents because of their clear
efficacy in the nondiabetic population. Since hypertension is often difficult
to control with a single agent (especially in type 2 DM), multiple
antihypertensive agents are usually required when blood pressure goals
(<130/85 mmHg) are not achieved. In this setting, long-acting calcium
channel antagonists should be considered as additional, or second-line, agents,
as these drugs appear to provide protection against cardiovascular events. ACE
inhibitors are contraindicated in pregnant diabetic patients and those
anticipating pregnancy. Because of the high prevalence of atherosclerotic
disease in individuals with DM, the possibility of renovascular hypertension
should be considered when the blood pressure is not readily controlled.
LOWER EXTREMITY COMPLICATIONS
DM
is the leading cause of nontraumatic lower extremity amputation in the United
States. Foot ulcers and infections are also a major source of morbidity in
individuals with DM. The reasons for the increased incidence of these disorders
in DM are complex and involve the interaction of several pathogenic factors:
neuropathy, abnormal foot biomechanics, peripheral vascular disease, and poor
wound healing. The peripheral sensory neuropathy interferes with normal
protective mechanisms and allows the patient to sustain major or repeated minor
trauma to the foot, often without knowledge of the injury. Disordered
proprioception causes abnormal weight bearing while walking and subsequent
formation of callus or ulceration. Motor and sensory neuropathy leads to
abnormal foot muscle mechanics and to structural changes in the foot (hammer
toe, claw toe deformity, prominent metatarsal heads). Autonomic neuropathy
results in anhidrosis and altered superficial blood flow in the foot, which
promote drying of the skin and fissure formation. Peripheral vascular disease
and poor wound healing impede resolution of minor breaks in the skin, allowing
them to enlarge and to become infected.
Approximately
15% of individuals with DM develop a foot ulcer, and a significant subset of
those individuals will at some time undergo amputation (14 to 24% risk with
that ulcer or subsequent ulceration). Risk factors for foot ulcers or
amputation include: male sex, diabetes >10 years' duration, peripheral
neuropathy, abnormal structure of foot (bony abnormalities, callus, thickened
nails), peripheral vascular disease, smoking, and history of previous ulcer or
amputation. Glycemic control is also a risk factor-each 2% increase in the
HbA1c increases the risk of a lower extremity ulcer by 1.6 times and the risk
of lower extremity amputation by 1.5 times.
![]() Treatment
Treatment
The
optimal therapy for foot ulcers and amputations is prevention through
identification of high-risk patients, education of the patient, and institution
of measures to prevent ulceration. High-risk patients should be identified
during the routine foot examination performed on all patients with DM (see
"Ongoing Aspects of Comprehensive Diabetes Care," below). Patient
education should emphasize: (1) careful selection of footwear, (2) daily
inspection of the feet to detect early signs of poor-fitting footwear or minor
trauma, (3) daily foot hygiene to keep the skin clean and moist, (4) avoidance
of self-treatment of foot abnormalities and high-risk behavior (e.g., walking barefoot),
and (5) prompt consultation with a health care provider if an abnormality
arises. Patients at high risk for ulceration or amputation may benefit from
evaluation by a foot care specialist. Interventions directed at risk factor
modification include orthotic shoes and devices, callus management, nail care,
and prophylactic measures to reduce increased skin pressure from abnormal bony
architecture. Attention to other risk factors for vascular disease (smoking,
dyslipidemia, hypertension) and improved glycemic control are also important.
Despite
preventive measures, foot ulceration and infection are common and represent a
potentially serious problem. Due to the multifactorial pathogenesis of lower
extremity ulcers, management of these lesions must be multidisciplinary and
often demands expertise in orthopedics, vascular surgery, endocrinology,
podiatry, and infectious diseases. The plantar surface of the foot is the most
common site of ulceration. Cellulitis without ulceration is also frequent and
should be treated with antibiotics that provide broad-spectrum coverage,
including anaerobes (see below).
An
infected ulcer is a clinical diagnosis, since superficial culture of any
ulceration will likely find multiple possible bacterial pathogens. The
infection surrounding the foot ulcer is often the result of multiple organisms
(gram-positive and -negative cocci and anaerobes), and gas gangrene may develop
in the absence of clostridial infection. Cultures taken from the debrided ulcer
base or from purulent drainage are most helpful. Wound depth should be
determined by inspection and probing with a blunt-tipped sterile instrument.
Plain radiographs of the foot should be performed to assess the possibility of
osteomyelitis in chronic ulcers that have not responded to therapy. Nuclear
medicine bone scans may be helpful, but overlying subcutaneous infection is
often difficult to distinguish from osteomyelitis. Indium-labeled white cell
studies are more useful in determining if the infection involves bony
structures or only soft tissue, but they are technically demanding. Magnetic
resonance imaging of the foot may be the most specific modality, although
distinguishing bony destruction due to osteomyelitis from destruction secondary
to Charcot arthropathy is difficult. If surgical debridement is necessary, bone
biopsy and culture usually provide the answer.
Osteomyelitis
is best treated by a combination of prolonged antibiotics and debridement of
infected bone. The possible contribution of vascular insufficiency should be
considered in all patients. Noninvasive blood-flow studies are often unreliable
in DM, and angiography may be required, recognizing the risk of
contrast-induced nephrotoxicity. Peripheral vascular bypass procedures are
often effective in promoting wound resolution and in decreasing the need for
amputation of the ischemic limb.
A
growing number of possible treatments for diabetic foot ulcers exist, but they
have yet to demonstrate clear efficacy in prospective, controlled trials. A
recent consensus statement from the ADA identified six interventions with
demonstrated efficacy in diabetic foot wounds: (1) off-loading, (2)
debridement, (3) wound dressings, (4) appropriate use of antibiotics, (5)
revascularization, and (6) limited amputation. Off-loading is the complete
avoidance of weight bearing on the ulcer, which removes the mechanical trauma
that retards wound healing. Bed rest and a variety of orthotic devices limit
weight bearing on wounds or pressure points. Surgical debridement of
neuropathic wounds is important and effective, but clear efficacy of other
modalities for wound cleaning (enzymes, soaking, whirlpools) is lacking.
Dressings promote wound healing by creating a moist environment and protecting
the wound. Antiseptic agents and topical antibiotics should be avoided.
Referral for physical therapy, orthotic evaluation, and rehabilitation may be
useful once the infection is controlled.
Mild
or non-limb-threatening infections can be treated with oral antibiotics
(cephalosporin, clindamycin, amoxicillin/clavulanate, and fluoroquinolones),
surgical debridement of necrotic tissue, local wound care (avoidance of weight
bearing over the ulcer), and close surveillance for progression of infection.
More severe ulcers may require intravenous antibiotics as well as bed rest and
local wound care. Urgent surgical debridement may be required. Intravenous
antibiotics should provide broad-spectrum coverage directed toward Staphylococcus
aureus, streptococci, gram-negative aerobes, and anaerobic bacteria.
Initial antimicrobial regimens include cefotetan, ampicillin/sulbactam, or the
combination of clindamycin and a fluoroquinolone. Severe infections, or
infections that do not improve after 48 h of antibiotic therapy, require
expansion of antimicrobial therapy to treat methicillin-resistant S. aureus
(e.g., vancomycin) and Pseudomonas aeruginosa. If the infection
surrounding the ulcer is not improving with intravenous antibiotics,
reassessment of antibiotic coverage and reconsideration of the need for
surgical debridement or revascularization are indicated. With clinical
improvement, oral antibiotics and local wound care can be continued on an
outpatient basis with close follow-up. As infection improves, a comprehensive
assessment of modifiable risk factors for foot ulceration should be performed
and should involve health professionals with expertise in podiatry, orthotics,
vascular surgery, and orthopedics.
New
information about wound biology has led to a number of new technologies (e.g.,
living skin equivalents and growth factors such as basic fibroblast growth
factor) that may prove useful. Recombinant platelet-derived growth factor has
some benefit and complements the basic therapies of off-loading, debridement,
and antibiotics. Hyperbaric oxygen has been used, but rigorous proof of
efficacy is lacking.
INFECTIONS
Individuals
with DM exhibit a greater frequency and severity of infection. The reasons for
this increase include incompletely defined abnormalities in cell-mediated
immunity and phagocyte function associated with hyperglycemia, as well as
diminished vascularization secondary to long-standing diabetes. Hyperglycemia
likely aids the colonization and growth of a variety of organisms (Candida
and other fungal species). Many common infections are more frequent and severe
in the diabetic population, whereas several rare infections are seen almost
exclusively in the diabetic population. Examples of this latter category
includes rhinocerebral mucormycosis and malignant otitis externa, which is
usually secondary to P. aeruginosa infection in the soft tissue
surrounding the external auditory canal. Malignant otitis externa begins with
pain and discharge and may progress rapidly to osteomyelitis and meningitis.
These infections should be sought, in particular, in patients presenting with
NKHS.
Pneumonia,
urinary tract infections, and skin and soft tissue infections are all more
common in the diabetic population. In general, the organisms that cause
pulmonary infections are similar to those found in the nondiabetic population;
however, gram-negative organisms, S. aureus, and Mycobacterium
tuberculosis are more frequent pathogens. Urinary tract infections (either
lower tract or pyelonephritis) are the result of common bacterial agents such
as Escherichia coli, though several yeast species (Candida and Torulopsis
glabrata) are commonly observed. Complications of urinary tract infections
include emphysematous pyelonephritis and emphysematous cystitis. Bacteriuria
occurs frequently in individuals with diabetic cystopathy. Susceptibility to
furunculosis, superficial candidal infections, and vulvovaginitis is increased.
Poor glycemic control is a common denominator in individuals with these
infections. Diabetic individuals have an increased rate of colonization of S.
aureus in the skin folds and nares. Diabetic patients also have a greater
risk of postoperative wound infections.
DERMATOLOGIC MANIFESTATIONS
The
most common skin manifestations of DM are protracted wound healing and skin
ulcerations. Diabetic dermopathy, sometimes termed pigmented pretibial
papules, or "diabetic skin spots," begins as an erythematous area
and evolves into an area of circular hyperpigmentation (Fig. 333-16). These
lesions result from minor mechanical trauma in the pretibial region and are
more common in elderly men with DM. Bullous diseases (shallow ulcerations or
erosions in the pretibial region) are also seen. Necrobiosis lipoidica
diabeticorum is a rare disorder of DM that predominantly affects young
women with type 1 DM, neuropathy, and retinopathy. It usually begins in the
pretibial region as an erythematous plaque or papules that gradually enlarge,
darken, and develop irregular margins, with atrophic centers and central
ulceration. They may be painful. Acanthosis nigricans (hyperpigmented
velvety plaques seen on the neck or extensor surfaces) is sometimes a feature
of severe insulin resistance and accompanying diabetes (Fig. 333-17).
Generalized or localized granuloma annulare (erythematous plaques on the
extremities or trunk) and scleredema (areas of skin thickening on the
back or neck at the site of previous superficial infections) are more common in
the diabetic population. Lipoatrophy and lipohypertrophy can
occur at insulin injection sites but are unusual with the use of human insulin.
Xerosis and pruritus are common and are relieved by skin moisturizers.
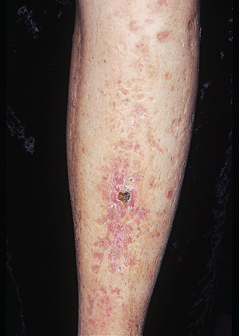
Figure 333-16: Diabetic dermopathy A crusted erosion at the
site of traumatic injury and many old pink depressed areas are seen on the
anterior leg of a 56-year-old male with diabetes mellitus. The other leg had
identical findings.
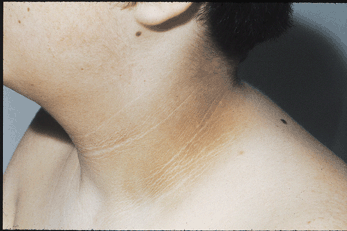
Figure 333-17: Acanthosis nigricans Velvety, dark-brown
epidermal thickening of the neck with prMninent skin fold andfeathered edges in
a 25-year-old obese woman with a family history of acanthosis nigricans.
Changes had been stable for more than five years. There were similar changes in
the axillae, antecubital fossae, and dorsum of the knuckles.
![]() Approach to
the Patient
Approach to
the Patient
DM
and its complications produce a wide range of symptoms and signs; those
secondary to acute hyperglycemia may occur at any stage of the disease, whereas
those related to chronic complications begin to appear during the second decade
of hyperglycemia. Individuals with previously undetected type 2 DM may present
with chronic complications of DM at the time of diagnosis. The history and
physical examination should assess for symptoms or signs of acute hyperglycemia
and should screen for the chronic complications and conditions associated with
DM.
History
A
complete medical history should be obtained with special emphasis on
DM-relevant aspects such as weight, family history of DM and its complications,
risk factors for cardiovascular disease, prior medical conditions, exercise,
smoking, and ethanol use. Symptoms of hyperglycemia include polyuria,
polydipsia, weight loss, fatigue, weakness, blurry vision, frequent superficial
infections (vaginitis, fungal skin infections), and slow healing of skin
lesions after minor trauma. Metabolic derangements relate mostly to
hyperglycemia (osmotic diuresis, reduced glucose entry into muscle) and to the
catabolic state of the patient (urinary loss of glucose and calories, muscle
breakdown due to protein degradation and decreased protein synthesis). Blurred
vision results from changes in the water content of the lens and resolves as
the hyperglycemia is controlled.
In
a patient with established DM, the initial assessment should also include
special emphasis on prior diabetes care, including the type of therapy, prior
HbA1c levels, self-monitoring blood glucose results, frequency of hypoglycemia,
presence of DM-specific complications, and assessment of the patient's
knowledge about diabetes. The chronic complications may afflict several organ
systems, and an individual patient may exhibit some, all, or none of the
symptoms related to the complications of DM (see above). In addition, the
presence of DM-related comorbidities should be sought (cardiovascular disease, hypertension,
dyslipidemia).
Physical
Examination
In
addition to a complete physical examination, special attention should be given
to DM-relevant aspects such as weight or body mass index, retinal examination,
orthostatic blood pressure, foot examination, peripheral pulses, and insulin
injection sites. Careful examination of the lower extremities should seek
evidence of peripheral neuropathy, calluses, superficial fungal infections,
nail disease, and foot deformities (such as hammer or claw toes and Charcot
foot) in order to identify sites of potential skin ulceration. Vibratory
sensation (128-MHz tuning fork at the base of the great toe) and the ability to
sense touch with a monofilament (5.07, 10-g monofilament) are useful to detect
moderately advanced diabetic neuropathy. Since dental disease is more frequent
in DM, the teeth and gums should also be examined.
Classification
of DM in an Individual Patient
The
etiology of diabetes in an individual with new-onset disease can usually be
assigned on the basis of clinical criteria. Individuals with type 1 DM tend to
have the following characteristics: (1) onset of disease prior to age 30; (2)
lean body habitus; (3) requirement of insulin as the initial therapy; (4)
propensity to develop ketoacidosis; and (5) an increased risk of other
autoimmune disorders such as autoimmune thyroid disease, adrenal insufficiency,
pernicious anemia, and vitiligo. In contrast, individuals with type 2 DM often
exhibit the following features: (1) develop diabetes after the age of 30; (2)
are usually obese (80% are obese, but elderly individuals may be lean); (3) may
not require insulin therapy initially; and (4) may have associated conditions
such as insulin resistance, hypertension, cardiovascular disease, dyslipidemia,
or polycystic ovary syndrome. In type 2 DM, insulin resistance is often
associated with abdominal obesity (as opposed to hip and thigh obesity) and
hypertriglyceridemia. Although most individuals diagnosed with type 2 DM are
older, the age of diagnosis appears to be declining in some ethnic groups, and
there is a marked increase among overweight teenagers. On the other hand, some
individuals (<10%) with the phenotypic appearance of type 2 DM do not have
absolute insulin deficiency but have autoimmune markers suggestive of type 1
DM. Thus, despite the revised classification of DM, it is remains difficult to
categorize some patients unequivocally. Individuals who deviate from the
clinical profile of type 1 and type 2 DM, or who have other associated defects
such as deafness, pancreatic exocrine disease, and other endocrine disorders,
should be classified accordingly (Table 333-1).
Laboratory
Assessment
The
laboratory assessment should first determine whether the patient meets the
diagnostic criteria for DM (Table 333-2) and should then assess the degree of
glycemic control (HbA1c, discussed below). In addition to the standard
laboratory evaluation, the patient should be screened for DM-associated
conditions (e.g., microalbuminuria, dyslipidemia, thyroid dysfunction). Individuals
at high risk for cardiovascular disease should be screened for asymptomatic
coronary artery disease by appropriate cardiac stress testing, when indicated.
The
classification of the type of DM does not usually require laboratory
assessments. Serum insulin or C-peptide measurements do not clearly distinguish
type 1 from type 2 DM at the time of diabetes onset; a low C-peptide level
merely confirms a patient's need for insulin. Conversely, many individuals with
new-onset type 1 DM retain some C-peptide production. Measurement of islet cell
antibodies at the time of diabetes onset may be useful if the type of DM is not
clear based on the characteristics discussed above, but this knowledge does not
usually alter therapy, which is based primarily on empirical metabolic
features.
LONG-TERM TREATMENT
OVERALL PRINCIPLES
The
goals of therapy for type 1 or type 2 DM are to: (1) eliminate symptoms related
to hyperglycemia, (2) reduce or eliminate the long-term microvascular and
macrovascular complications of DM, and (3) allow the patient to achieve as
normal a life-style as possible. To reach these goals, the physician should
identify a target level of glycemic control for each patient, provide the
patient with the educational and pharmacologic resources necessary to reach
this level, and monitor/treat DM-related complications. Symptoms of diabetes
usually resolve when the plasma glucose is <11.1 mmol/L (200 mg/dL), and
thus most DM treatment focuses on achieving the second and third goals.
The
care of an individual with either type 1 or type 2 DM requires a
multidisciplinary team. Central to the success of this team are the patient's
participation, input, and enthusiasm, all of which are essential for optimal
diabetes management. Members of the health care team include the primary care
provider and/or the endocrinologist or diabetologist, a certified diabetes
educator, and a nutritionist. In addition, when the complications of DM arise,
subspecialists (including neurologists, nephrologists, vascular surgeons,
cardiologists, ophthalmologists, and podiatrists) with experience in DM-related
complications are essential.
A
number of names are sometimes applied to different approaches to diabetes care,
such as intensive insulin therapy, intensive glycemic control, and "tight
control." The current chapter, however, will use the term comprehensive
diabetes care to emphasize the fact that optimal diabetes therapy involves
more than plasma glucose management. Though glycemic control is central to
optimal diabetes therapy, comprehensive diabetes care of both type 1 and type 2
DM should also detect and manage DM-specific complications and modify risk
factors for DM-associated diseases.
In
addition to assessing the physical aspects of the patient with DM, the
physician and members of the diabetes management team should consider social,
family, financial, cultural, and employment-related issues that may have an
impact on diabetes care. With this information, the physician can work with the
patient and his or her family to establish therapeutic goals and design a
comprehensive and feasible plan for optimal diabetes care.
EDUCATION OF THE PATIENT ABOUT DM, NUTRITION,
AND EXERCISE
Patient
participation is an essential component of comprehensive diabetes care. The
patient with type 1 or type 2 DM should receive education about nutrition,
exercise, care of diabetes during illness, and medications to lower the plasma
glucose. Along with improved compliance, patient education allows individuals
with DM to assume greater responsibility for their care. Patient education
should be viewed as a continuing process with regular visits for reinforcement;
it should not be a process that is completed after one or two visits to
a nurse educator or nutritionist.
Diabetes Education
The
diabetes educator is a health care professional (nurse, dietician, or
pharmacist) with specialized patient education skills who is certified in
diabetes education (indicating demonstrated skills in diabetes knowledge and
education and certification by the American Association of Diabetes Educators).
The educator is a vital member of the comprehensive diabetes care program and
educates the patient about a number of issues important for optimal diabetes
care, including self-monitoring of blood glucose; urine ketone monitoring (type
1 DM); insulin administration; guidelines for diabetes management during
illnesses; management of hypoglycemia; foot and skin care; diabetes management
before, during, and after exercise; and risk factor-modifying activities.
Nutrition
Medical nutrition therapy (MNT) is a term used by the ADA to
describe the optimal coordination of caloric intake with other aspects of
diabetes therapy (insulin, exercise, weight loss). Historically, nutrition has
imposed restrictive, complicated regimens on the patient. Current practices
have greatly changed, though many patients and health care providers still view
the diabetic diet as monolithic and static. For example, modern MNT now
includes foods with sucrose and seeks to modify other risk factors such as
hyperlipidemia and hypertension rather than focusing exclusively on weight loss
in individuals with type 2 DM. Like other aspects of DM therapy, MNT must be
adjusted to meet the goals of the individual patient. Furthermore, MNT
education is an important component of comprehensive diabetes care and should
be reinforced by regular patient education. In general, the components of
optimal MNT are similar for individuals with type 1 or type 2 DM (Table 333-8).
Table 333-8: Nutritional Recommendations for All
Persons with Diabetes
|
|||
|
|
||
The
goal of MNT in the individual with type 1 DM is to coordinate and match the
caloric intake, both temporally and quantitatively, with the appropriate amount
of insulin. MNT in type 1 DM and self-monitoring of blood glucose must be
integrated to define the optimal insulin regimen. MNT must be flexible enough
to allow for exercise, and the insulin regimen must allow for deviations in
caloric intake. An important component of MNT in type 1 DM is to minimize the
weight gain often associated with intensive diabetes management.
The
goals of MNT in type 2 DM are slightly different and address the greatly
increased prevalence of cardiovascular risk factors (hypertension,
dyslipidemia, obesity) and disease in this population. The majority of these
individuals are obese, and weight loss is still strongly encouraged and should
remain an important goal. Medical treatment of obesity is a rapidly evolving
area and is discussed in Chap. 77. Hypocaloric diets and modest weight loss
often result in rapid and dramatic glucose lowering in individuals with
new-onset type 2 DM. Nevertheless, numerous studies document that long-term
weight loss is uncommon. Therefore, current MNT for type 2 DM should emphasize
modest caloric reduction, increased physical activity, and reduction of
hyperlipidemia and hypertension. Increased consumption of soluble, dietary
fiber may improve glycemic control in individuals with type 2 DM.
Exercise
Exercise
is an integral component of comprehensive diabetes care that can have multiple
positive benefits (cardiovascular benefits, reduced blood pressure, maintenance
of muscle mass, reduction in body fat, weight loss, etc.). For individuals with
type 1 or type 2 DM, exercise is also useful for lowering plasma glucose
(during and following exercise) and increasing insulin sensitivity.
Despite
its benefits, exercise presents several challenges for individuals with DM
because they lack the normal glucoregulatory mechanisms. Skeletal muscle is a
major site for metabolic fuel consumption in the resting state, and the
increased muscle activity during vigorous, aerobic exercise greatly increases
fuel requirements. Individuals with type 1 DM are prone to either hyperglycemia
or hypoglycemia during exercise, depending on the preexercise plasma glucose,
the circulating insulin level, and the level of exercise-induced
catecholamines. If the insulin level is too low, the rise in catecholamines may
increase the plasma glucose excessively, promote ketone body formation, and
possibly lead to ketoacidosis. Conversely, if the circulating insulin level is
excessive, this relative hyperinsulinemia may reduce hepatic glucose production
(decreased glycogenolysis, decreased gluconeogenesis) and increase glucose entry
into muscle, leading to hypoglycemia.
To
avoid exercise-related hyper- or hypoglycemia, individuals with type 1 DM
should: (1) monitor blood glucose before, during, and after exercise; (2) delay
exercise if blood glucose is >14 mmol/L (250 mg/dL), <5.5 mmol/L (100
mg/dL), or if ketones are present; (3) eat a meal 1 to 3 h before exercise and
take supplemental carbohydrate feedings at least every 30 min during vigorous
or prolonged exercise; (4) decrease insulin doses (based on previous
experience) before exercise and inject insulin into a nonexercising area; and
(5) learn individual glucose responses to different types of exercise and
increase food intake for up to 24 h after exercise, depending on intensity and
duration of exercise. In individuals with type 2 DM, exercise-related
hypoglycemia is less common but can occur in individuals taking either insulin
or sulfonylureas.
Because
asymptomatic cardiovascular disease appears at a younger age in both type 1 and
type 2 DM, formal exercise tolerance testing may be warranted in diabetic
individuals with any of the following: age ![]() 35
years, long-standing type 1 DM (>20 to 25 years' duration), microvascular
complications of DM (retinopathy, microalbuminuria, or nephropathy), peripheral
vascular disease, other risk factors of coronary artery disease, or autonomic
neuropathy. Untreated proliferative retinopathy is a relative contraindication
to vigorous exercise, since this may lead to vitreous hemorrhage or retinal
detachment.
35
years, long-standing type 1 DM (>20 to 25 years' duration), microvascular
complications of DM (retinopathy, microalbuminuria, or nephropathy), peripheral
vascular disease, other risk factors of coronary artery disease, or autonomic
neuropathy. Untreated proliferative retinopathy is a relative contraindication
to vigorous exercise, since this may lead to vitreous hemorrhage or retinal
detachment.
MONITORING THE LEVEL OF GLYCEMIC CONTROL
Optimal
monitoring of glycemic control involves plasma glucose measurements by the
patient and an assessment of long-term control by the physician (measurement of
HbA1c and review of the patient's self-measurements of plasma glucose). These
measurements are complementary: the patient's measurements provide a picture of
short-term glycemic control, whereas the HbA1c reflects average glycemic
control over the previous 2 to 3 months. Integration of both measurements
provides an accurate assessment of the glycemic control achieved.
Self-Monitoring of Blood Glucose
Self-monitoring
of blood glucose (SMBG) is the standard of care in diabetes management and
allows the patient to monitor his or her blood glucose at any time. In SMBG, a
small drop of blood and an easily detectable enzymatic reaction allow
measurement of the capillary plasma glucose. By combining glucose measurements
with diet history, medication changes, and exercise history, the physician and
patient can improve the treatment program.
The
frequency of SMBG measurements must be individualized and adapted to address
the goals of diabetes care as defined by the patient and the health care
provider. Individuals with type 1 DM should routinely measure their plasma
glucose four to eight times per day to estimate and select mealtime boluses of
short-acting insulin and to modify long-acting insulin doses. Most individuals
with type 2 DM require less frequent monitoring, though the optimal frequency
of SMBG has not been clearly defined. Individuals with type 2 DM who are on
oral medications should utilize SMBG as a means of assessing the efficacy of
their medication and diet. Since plasma glucose levels fluctuate less in these
individuals, one to two SMBG measurements per day (or fewer) may be sufficient.
Individuals with type 2 DM who are on insulin should utilize SMBG more
frequently than those on oral agents.
Two
devices for continuous blood glucose monitoring have been recently approved by
the U.S. Food and Drug Administration (FDA). The Glucowatch uses iontophoresis
to assess glucose in interstitial fluid, whereas the Minimed device uses an
indwelling subcutaneous catheter to monitor interstitial fluid glucose. Both
devices utilize immobilized glucose oxidase to generate electrons in response
to changing glucose levels. Though clinical experience with these devices is
limited, they perform well in clinical trials and appear to provide useful
short-term information about the patterns of glucose changes as well as an
enhanced ability to detect hypoglycemic episodes.
Although
urine glucose testing does not provide an accurate assessment of glycemic
control, urine ketones are a sensitive indicator of early diabetic ketoacidosis
and should be measured in individuals with type 1 DM when the plasma glucose is
consistently >16.7 mmol/L (300 mg/dL); during a concurrent illness; or with
symptoms such as nausea, vomiting, or abdominal pain.
Assessment of Long-Term Glycemic Control
Measurement
of glycated hemoglobin is the standard method for assessing long-term glycemic
control. When plasma glucose is consistently elevated, there is an increase in
nonenzymatic glycation of hemoglobin; this alteration reflects the glycemic
history over the previous 2 to 3 months, since erythrocytes have an average
life span of 120 days. There are numerous laboratory methods for measuring the
various forms of glycated hemoglobin, and these have significant interassay
variations. Because of its superior specificity and reliability, the HbA1c
assay performed by the high-performance liquid chromatography (HPLC) method has
become the standard reference method for most glycated hemoglobin measurements.
Since glycated hemoglobin measurements are usually compared to prior
measurements, it is essential for the assay results to be comparable. Depending
on the assay methodology for HbA1c, hemoglobinopathies, hemolytic anemia, and
uremia may interfere with the HbA1c result.
Glycated
hemoglobin or HbA1c should be measured in all individuals with DM during their
initial evaluation and as part of their comprehensive diabetes care. As the
primary predictor of long-term complications of DM, the HbA1c should mirror, to
a certain extent, the short-term measurements of SMBG. These two measurements
are complementary in that recent intercurrent illnesses may impact the SMBG
measurements but not the HbA1c. Likewise, postprandial and nocturnal
hyperglycemia may not be detected by the SMBG of fasting and preprandial
capillary plasma glucose but will be reflected in the HbA1c. When measured by
HPLC, the HbA1c approximates the following mean plasma glucose values: an HbA1c
of 6% is 6.6 mmol/L (120 mg/dL), 7% is 8.3 mmol/L (150 mg/dL), 8% is 10.0
mmol/L (180 mg/dL), etc. [A 1% rise in the HbA1c translates into a 1.7-mmol/L
(30 mg/dL) increase in the mean glucose.] The degree of glycation of other
proteins, such as albumin, has been used as an alternative indicator of
glycemic control when the HbA1c is inaccurate (hemolytic anemia,
hemoglobinopathies). The fructosamine assay (using albumin) is an example of an
alternative measurement of glycemic control and reflects the glycemic status
over the 2 to 4 prior weeks. Current consensus statements do not favor the use
of alternative assays of glycemic control, as there are no studies to indicate
whether such assays accurately predict the complications of DM.
![]() Treatment
Treatment
Establishment of a Target Level of Glycemic
Control
Because
the complications of DM are related to glycemic control, normoglycemia or near
normoglycemia is the desired, but often elusive, goal for most patients.
However, normalization of the plasma glucose for long periods of time is
extremely difficult, as demonstrated by the DCCT. Regardless of the level of
hyperglycemia, improvement in glycemic control will lower the risk of diabetes
complications (Fig. 333-8).
The
target for glycemic control (as reflected by the HbA1c) must be individualized,
and the health care provider should establish the goals of therapy in
consultation with the patient after considering a number of medical, social,
and life-style issues. Some important factors to consider include the patient's
age, ability to understand and implement a complex treatment regimen, presence
and severity of complications of diabetes, ability to recognize hypoglycemic
symptoms, presence of other medical conditions or treatments that might alter
the response to therapy, life-style and occupation (e.g., possible consequences
of experiencing hypoglycemia on the job), and level of support available from
family and friends.
The
ADA has established suggested glycemic goals based on the premise that glycemic
control predicts development of DM-related complications. In general, the
target HbA1c should be <7.0% (Table 333-9). Other consensus groups (such as
the Veterans Administration) have suggested HbA1c goals that take into account
the patient's life expectancy at the time of diagnosis and the presence of
microvascular complications. Such recommendations strive to balance the
financial and personal costs of glycemic therapy with anticipated benefits
(reduced health care costs, reduced morbidity). One limitation to this approach
is that the onset of hyperglycemia in type 2 DM is difficult to ascertain and
likely predates the diagnosis. Furthermore, though the life expectancy can be
predicted for a patient population, the physician must treat an individual
patient; consequently, the target HbA1c must be individualized to accommodate
these other considerations.
Table 333-9: Ideal Goals for Glycemic Controla
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Type 1 Diabetes Mellitus
General Aspects
Comprehensive
diabetes care should be instituted in all individuals with type 1 DM and should
involve attention to nutrition, exercise, and risk factor management in
addition to insulin administration. The ADA recommendations for fasting and
bedtime glycemic goals and HbA1c targets are summarized in Table 333-9. The
goal is to design and implement insulin regimens that mimic physiologic insulin
secretion. Because individuals with type 1 DM lack endogenous insulin
production, administration of basal, exogenous insulin is essential for
regulating glycogen breakdown, gluconeogenesis, lipolysis, and ketogenesis.
Likewise, postprandial insulin replacement should be appropriate for the
carbohydrate intake and promote normal glucose utilization and storage.
Intensive Management
Intensive
diabetes management is defined by the ADA as "…a mode of treatment for the
person with DM that has the goal of achieving euglycemia or near-normal
glycemia using all available resources to accomplish this goal." These
resources include thorough and continuing patient education, comprehensive
recording of plasma glucose measurements and nutrition intake by the patient,
and a variable insulin regimen that matches glucose intake and insulin dose.
Insulin regimens usually include multiple-component insulin regimens, multiple
daily injections (MDI), or insulin infusion devices (all discussed below).
The
benefits of intensive diabetes management and improved glycemic control include
a reduction in the microvascular complications of DM and a possible delay or
reduction in the macrovascular complications of DM. From a psychological
standpoint, the patient experiences greater control over his or her diabetes
and often notes an improved sense of well-being, greater flexibility in the
timing and content of meals, and the capability to alter insulin dosing with
exercise. In addition, intensive diabetes management in pregnancy reduces fetal
malformation and morbidity. Intensive diabetes management is also strongly
encouraged in newly diagnosed patients with type 1 DM because it may prolong
the period of C-peptide production, which may result in better glycemic control
and a reduced risk of serious hypoglycemia.
Although
intensive management confers impressive benefits, it is also accompanied by
significant personal and financial costs and is therefore not appropriate for
all individuals. It requires a combination of dedication, persistence, and
motivation on the part of the patient, as well as medical, educational,
nursing, nutritional, and psychological expertise on the part of the diabetes
management team. Circumstances in which intensive diabetes management should be
strongly considered are listed in Table 333-10.
Table 333-10: Indications for Intensive Diabetes
Management
|
Insulin Preparations
Current
insulin preparations are generated by recombinant DNA technology and consist of
the amino acid sequence of human insulin. Animal insulin (beef or pork) is no
longer used. Human insulin has been formulated with distinctive
pharmacokinetics to mimic physiologic insulin secretion (Table 333-11). In the
United States, all insulin is formulated as U-100 (100 units/mL), whereas in
some other countries it is available in other units (e.g., U-40 = 40 units/mL).
One short-acting insulin formulation, lispro, is an insulin analogue in which
the 28th and 29th amino acids (lysine and proline) on the insulin B chain have
been reversed by recombinant DNA technology. This insulin analogue has full
biologic activity but less tendency toward subcutaneous aggregation, resulting
in more rapid absorption and onset of action and a shorter duration of action.
These characteristics are particularly advantageous for allowing entrainment of
insulin injection and action to rising plasma glucose levels following meals,
although improvement in HbA1c values have not been found consistently. The
shorter duration of action also appears to be associated with a decreased
number of hypoglycemic episodes, primarily because the decay of lispro action
corresponds better to the decline in plasma glucose after a meal. Insulin
glargine is a long-acting biosynthetic human insulin that differs from normal
insulin in that asparagine is replaced by glycine at amino acid 21, and two
arginine residues are added to the C-terminus of the B chain. Compared to NPH
insulin, the onset of insulin glargine action is later, the duration of action
is longer (~24 h), and there is no pronounced peak. A lower incidence of
hypoglycemia, especially at night, was reported in one trial with insulin
glargine when compared to NPH insulin. Since glargine has only recently
approved, clinical experience is limited. Additional insulin analogues are
currently under development.
Table 333-11: Pharmacokinetics of Insulin
Preparations
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Basal
insulin requirements are provided by intermediate (NPH or lente) or long-acting
(ultralente or glargine) insulin formulations. These are usually combined with
short-acting insulin in an attempt to mimic physiologic insulin release with
meals. Although mixing of intermediate and short-acting insulin formulations is
common practice, this mixing may alter the insulin absorption profile
(especially those of short-acting insulins). For example, the absorption of
regular insulin is delayed when mixed for even short periods of time (<5
min) with lente or ultralente insulin, but not when mixed with NPH insulin.
Lispro absorption is delayed by mixing with NPH but not ultralente. Insulin
glargine should not be mixed with other insulins. The miscibility of human
regular and NPH insulin allows for the production of combination insulins that
contain 75% NPH and 25% regular (75/25), 70% NPH and 30% regular (70/30), or
equal mixtures of NPH and regular. These combinations of insulin are more
convenient for the patient but prevent adjustment of only one component of the
insulin formulation. The alteration in insulin absorption when the patient
mixes different insulin formulation should not discourage the patient from
mixing insulin. However, the following guidelines should be followed: (1) mix
the different insulin formulations in the syringe immediately before injection
(inject within 2 min after mixing); (2) if possible, do not store insulin as a
mixture; and (3) follow the same routine in terms of insulin mixing and
administration to standardize the physiologic response to injected insulin.
Insulin Regimens
Representations
of the various insulin regimens that may be utilized in type 1 DM are
illustrated in Fig. 333-12. Although the insulin profiles are depicted as
"smooth," symmetric curves, there is considerable patient-to-patient
variation in the peak and duration. In all regimens, long-acting insulins (NPH,
lente, ultralente, or glargine insulin) supply basal insulin, whereas prandial
insulin is provided by either regular or lispro insulin. Lispro should be
injected just before a meal; regular insulin is given 30 to 45 min prior to a
meal.
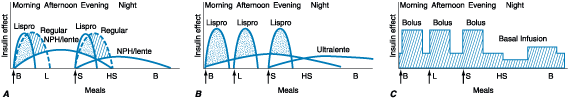
Figure 333-12: Representative insulin
regimens for the treatment of diabetes. For each panel, the y-axis shows the
amount of insulin effect and the x-axis shows the time of day. B, breakfast; L,
lunch; S, supper; HS, bedtime; CSII, continuous subcutaneous insulin infusion.
The time of insulin injection is shown with a vertical arrow. The type of
insulin is noted above each insulin curve. A. The injection of two shots
of intermediate-acting insulin (NPH or lente) and short-acting insulin (lispro
or regular). Only one formulation of short-acting insulin is used. B: A
multiple-component insulin regimen consisting of two shots of ultralente each
day to provide basal insulin coverage and three shots of Lispro to provide
glycemic coverage for each meal. The ultralente doses are usually 10 to 12 h
apart. C: Insulin administration by insulin infusion device is shown
with the basal insulin and a bolus injection at each meal. The basal insulin
rate is decreased during the evening and increased slightly prior to the
patient awakening in the morning.(Adapted from Intensive Diabetes Management,
2d ed, R. Farkas-Hirsch (ed). Alexandria, VA, American Diabetes Association,
1998)
A
shortcoming of current insulin regimens is that injected insulin immediately
enters the systemic circulation, whereas endogenous insulin is secreted into
the portal vein. Thus, exogenous insulin administration exposes the liver to
subphysiologic insulin levels. No insulin regimen reproduces the precise
insulin secretory pattern of the pancreatic islet. However, the most
physiologic regimens entail more frequent insulin injections, greater reliance
on short-acting insulin, and more frequent capillary plasma glucose
measurements. In general, individuals with type 1 DM require 0.5 to 1.0 U/kg
per day of insulin divided into multiple doses. Initial insulin-dosing regimens
should be conservative; approximately 40 to 50% of the insulin should be given
as basal insulin. A single daily injection of insulin is not appropriate
therapy in type 1 DM.
One
commonly used regimen consists of twice-daily injections of an intermediate
insulin (NPH or lente) mixed with a short-acting insulin before the morning and
evening meal (Fig. 333-12A). Such regimens usually prescribe two-thirds
of the total daily insulin dose in the morning (with about two-thirds given as
intermediate-acting insulin and one-third as short-acting) and one-third before
the evening meal (with approximately one-half given as intermediate-acting
insulin and one-half as short-acting). The drawback to such a regimen is that
it enforces a rigid schedule on the patient, in terms of daily activity and the
content and timing of meals. Although it is simple and effective at avoiding
severe hyperglycemia, it does not generate near-normal glycemic control in most
individuals with type 1 DM. Moreover, if the patient's meal pattern or content
varies or if physical activity is increased, hyperglycemia or hypoglycemia may
result. Moving the intermediate insulin from before the evening meal to bedtime
may avoid nocturnal hypoglycemia and provide more insulin as glucose levels
rise in the early morning (so-called dawn phenomenon). The insulin dose in such
regimens should be adjusted based on SMBG results with the following general
assumptions: (1) the fasting glucose is primarily determined by the prior
evening intermediate-acting insulin; (2) the pre-lunch glucose is a function of
the morning short-acting insulin; (3) the pre-supper glucose is a function of
the morning intermediate-acting insulin; and (4) the bedtime glucose is a
function of the pre-supper, short-acting insulin.
Multiple-component
insulin regimens refer to the combination of basal insulin; preprandial
short-acting insulin; and changes in short-acting insulin doses to accommodate
the results of frequent SMBG, anticipated food intake, and physical activity.
Sometimes also referred to as multiple daily injections, such regimens
offer the patient maximal flexibility in terms of life-style and the best
chance for achieving near normoglycemia. One such regimen, shown in Fig. 333-12B,
consists of a basal insulin with ultralente twice a day and preprandial lispro.
The lispro dose is based on individualized algorithms that integrate the
preprandial glucose and the anticipated carbohydrate intake. An alternative
multiple-component insulin regimen consists of bedtime intermediate insulin, a
small dose of intermediate insulin at breakfast (20 to 30% of bedtime dose),
and preprandial short-acting insulin. There are numerous variations of these
regimens that can be optimized for individual patients. Frequent SMBG (four to
8 times per day) is absolutely essential for these types of insulin regimens.
Continuous
subcutaneous insulin infusion (CSII) is another multiple-component insulin
regimen (Fig. 333-12C). Sophisticated insulin infusion devices are now
available that can accurately deliver small doses of insulin (microliters per
hour). For example, multiple basal infusion rates can be programmed to: (1)
accommodate nocturnal versus daytime basal insulin requirement, (2) alter
infusion rate during periods of exercise, or (3) select different waveforms of
insulin infusion. A preprandial insulin ("bolus") is delivered by the
insulin infusion device based on instructions from the patient, which follow
individualized algorithms that account for preprandial plasma glucose and
anticipated carbohydrate intake. These devices require a health professional
with considerable experience with insulin infusion devices and very frequent
patient interactions with the diabetes management team. Insulin infusion
devices present unique challenges, such as infection at the infusion site,
unexplained hyperglycemia because the infusion set becomes obstructed, or
diabetic ketoacidosis if the pump becomes disconnected. Since most physicians
use lispro insulin in CSII, the extremely short half-life of this insulin
quickly leads to insulin deficiency if the delivery system is interrupted.
Essential to the safe use of infusion devices is thorough patient education
about pump function and frequent SMBG.
Type 2 Diabetes Mellitus
General Aspects
The
goals of therapy for type 2 DM are similar to those in type 1: improved
glycemic control with near normalization of the HbA1c. While glycemic control
tends to dominate the management of type 1 DM, the care of individuals with
type 2 DM must also include attention to the treatment of conditions associated
with type 2 DM (obesity, hypertension, dyslipidemia, cardiovascular disease)
and detection/management of DM-related complications (Fig. 333-13). DM-specific
complications may be present in up to 20 to 50% of individuals with newly
diagnosed type 2 DM. Reduction in cardiovascular risk is of paramount
importance as this is the leading cause of mortality in these individuals.
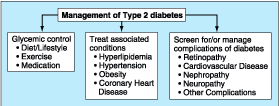
Figure 333-13: Essential elements in
comprehensive diabetes care of type 2 diabetes.
Diabetes
management should begin with MNT (discussed above). An exercise regimen to
increase insulin sensitivity and promote weight loss should also be instituted.
After MNT and increased physical activity have been instituted, glycemic
control should be reassessed; if the patient's glycemic target is not achieved
after 3 to 4 weeks of MNT, pharmacologic therapy is indicated. Pharmacologic
approaches to the management of type 2 DM include both oral glucose-lowering
agents and insulin; most physicians and patients prefer oral glucose-lowering
agents as the initial choice. Any therapy that improves glycemic control
reduces "glucose toxicity" to the islet cells and improves endogenous
insulin secretion.
Glucose-Lowering Agents
Recent
advances in the therapy of type 2 DM have generated considerable enthusiasm for
oral glucose-lowering agents that target different pathophysiologic processes
in type 2 DM. Based on their mechanisms of action, oral glucose-lowering agents
are subdivided into agents that increase insulin secretion, reduce glucose
production, or increase insulin sensitivity (Table 333-12). Oral
glucose-lowering agents (with the exception of ![]() -glucosidase
inhibitors) are ineffective in type 1 DM and should not be used for glucose
management of severely ill individuals with type 2 DM. Insulin is sometimes the
initial glucose-lowering agent.
-glucosidase
inhibitors) are ineffective in type 1 DM and should not be used for glucose
management of severely ill individuals with type 2 DM. Insulin is sometimes the
initial glucose-lowering agent.
Table 333-12: Oral Glucose-Lowering Therapies in
Type 2 DM
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Insulin Secretagogues
Insulin
secretagogues stimulate insulin secretion by interacting with the ATP-sensitive
potassium channel on the beta cell (Fig. 333-1). These drugs are most effective
in individuals with type 2 DM of relatively recent onset (<5 years), who
have endogenous insulin production and tend to be obese. At maximum doses,
first-generation sulfonylureas are similar in potency to second-generation
agents but have a longer half-life, a greater incidence of hypoglycemia, and
more frequent drug interactions (Table 333-13). Thus, second-generation
sulfonylureas are generally preferred. An advantage to a more rapid onset of
action is better coverage of the postprandial glucose rise, but the shorter
half-life of such agents requires more than once-a-day dosing. Sulfonylureas
reduce both fasting and postprandial glucose and should be initiated at low
doses and increased at 1- to 2-week intervals based on SMBG. In general,
sulfonylureas increase insulin acutely and thus should be taken shortly before
a meal; with chronic therapy, though, the insulin release is more sustained.
Replaglinide is not a sulfonylurea but also interacts with the ATP-sensitive
potassium channel. Because of its short half-life, it is usually given with or
immediately before each meal to reduce meal-related glucose excursions.
Table 333-13: Characteristics of Agents that
Increase Insulin Secretion
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Insulin
secretagogues are well tolerated in general. All of these agents, however, have
the potential to cause profound and persistent hypoglycemia, especially in
elderly individuals. Hypoglycemia is usually related to delayed meals,
increased physical activity, alcohol intake, or renal insufficiency.
Individuals who ingest an overdose of these agents develop prolonged and
serious hypoglycemia and should be monitored closely in the hospital (Chap.
334). Most sulfonylureas are metabolized in the liver to compounds that are
cleared by the kidney. Thus, their use in individuals with significant hepatic
or renal dysfunction is not advisable. Weight gain, a common side effect of
sulfonylurea therapy, results from the increased insulin levels and improvement
in glycemic control. Some sulfonylureas have significant drug interactions with
other medications such as alcohol, warfarin, aspirin, ketoconazole, ![]() -glucosidase
inhibitors, and fluconazole. Despite prior concerns that use of sulfonylureas
might increase cardiovascular risk, recent trials have refuted this claim.
-glucosidase
inhibitors, and fluconazole. Despite prior concerns that use of sulfonylureas
might increase cardiovascular risk, recent trials have refuted this claim.
Biguanides
Metformin
is representative of this class of agents. It reduces hepatic glucose
production through an undefined mechanism and may improve peripheral glucose
utilization slightly (Table 333-12). Metformin reduces fasting plasma glucose
and insulin levels, improves the lipid profile, and promotes modest weight
loss. The initial starting dose of 500 mg once or twice a day can be increased
to 850 mg tid or 1000 mg bid. Because of its relatively slow onset of action
and gastrointestinal symptoms with higher doses, the dose should be escalated
every 2 to 3 weeks based on SMBG measurements. The major toxicity of metformin,
lactic acidosis, can be prevented by careful patient selection. Metformin should
not be used in patients with renal insufficiency [serum creatinine >133 ![]() mol/L
(1.5 mg/dL) in men or >124
mol/L
(1.5 mg/dL) in men or >124 ![]() mol/L
(1.4 mg/dL) in women, with adjustments for age], any form of acidosis,
congestive heart failure, liver disease, or severe hypoxia. Metformin should be
discontinued in patients who are seriously ill, in patients who can take
nothing orally, and in those receiving radiographic contrast material. Insulin
should be used until metformin can be restarted. Though well tolerated in general,
some individuals develop gastrointestinal side effects (diarrhea, anorexia,
nausea, and metallic taste) that can be minimized by gradual dose escalation.
Because the drug is metabolized in the liver, it should not be used in patients
with liver disease or heavy ethanol intake.
mol/L
(1.4 mg/dL) in women, with adjustments for age], any form of acidosis,
congestive heart failure, liver disease, or severe hypoxia. Metformin should be
discontinued in patients who are seriously ill, in patients who can take
nothing orally, and in those receiving radiographic contrast material. Insulin
should be used until metformin can be restarted. Though well tolerated in general,
some individuals develop gastrointestinal side effects (diarrhea, anorexia,
nausea, and metallic taste) that can be minimized by gradual dose escalation.
Because the drug is metabolized in the liver, it should not be used in patients
with liver disease or heavy ethanol intake.
![]() ->nm><<016>>Glucosidase
inhibitors
->nm><<016>>Glucosidase
inhibitors
![]() -Glucosidase
inhibitors (acarbose and miglitol) reduce postprandial hyperglycemia by
delaying glucose absorption; they do not affect glucose utilization or insulin
secretion (Table 333-12). Postprandial hyperglycemia, secondary to impaired
hepatic and peripheral glucose disposal, contributes significantly to the
hyperglycemic state in type 2 DM. These drugs, taken just before each meal,
reduce glucose absorption by inhibiting the enzyme that cleaves
oligosaccharides into simple sugars in the intestinal lumen. Therapy should be
initiated at a low dose (25 mg of acarbose or miglitol) with the evening meal
and may be increased to a maximal dose over weeks to months (50 to 100 mg for
acarbose or 50 mg for miglitol with each meal). The major side effects
(diarrhea, flatulence, abdominal distention) are related to increased delivery
of oligosaccharides to the large bowel and can be reduced somewhat by gradual
upward dose titration.
-Glucosidase
inhibitors (acarbose and miglitol) reduce postprandial hyperglycemia by
delaying glucose absorption; they do not affect glucose utilization or insulin
secretion (Table 333-12). Postprandial hyperglycemia, secondary to impaired
hepatic and peripheral glucose disposal, contributes significantly to the
hyperglycemic state in type 2 DM. These drugs, taken just before each meal,
reduce glucose absorption by inhibiting the enzyme that cleaves
oligosaccharides into simple sugars in the intestinal lumen. Therapy should be
initiated at a low dose (25 mg of acarbose or miglitol) with the evening meal
and may be increased to a maximal dose over weeks to months (50 to 100 mg for
acarbose or 50 mg for miglitol with each meal). The major side effects
(diarrhea, flatulence, abdominal distention) are related to increased delivery
of oligosaccharides to the large bowel and can be reduced somewhat by gradual
upward dose titration. ![]() -Glucosidase
inhibitors may increase levels of sulfonylureas and increase the incidence of
hypoglycemia. Simultaneous treatment with bile acid resins and antacids should
be avoided. These agents should not be used in individuals with inflammatory
bowel disease, gastroparesis, or a serum creatinine >177
-Glucosidase
inhibitors may increase levels of sulfonylureas and increase the incidence of
hypoglycemia. Simultaneous treatment with bile acid resins and antacids should
be avoided. These agents should not be used in individuals with inflammatory
bowel disease, gastroparesis, or a serum creatinine >177 ![]() mol/L
(2.0 mg/dL). This class of agents is not as potent as other oral agents in
lowering the HbA1c but is unique in that it reduces the postprandial glucose
rise even in individuals with type 1 DM.
mol/L
(2.0 mg/dL). This class of agents is not as potent as other oral agents in
lowering the HbA1c but is unique in that it reduces the postprandial glucose
rise even in individuals with type 1 DM.
Thiazolidinediones
Thiazolidinediones
represent a new class of agents that reduce insulin resistance. These drugs
bind to a nuclear receptor (peroxisome proliferator-activated receptor, PPAR-![]() )
that regulates gene transcription. The PPAR-
)
that regulates gene transcription. The PPAR-![]() receptor is found at highest levels in adipocytes but is expressed at lower
levels in many other insulin-sensitive tissues. Agonists of this receptor
promote adipocyte differentiation and may reduce insulin resistance in skeletal
muscle indirectly. Thiazolidinediones reduce the fasting plasma glucose by
improving peripheral glucose utilization and insulin sensitivity (Table
333-12). Circulating insulin levels decrease with use of the
thiazolidinediones, indicating a reduction in insulin resistance. Although
direct comparisons are not available, the two currently available
thiazolidinediones appear to have similar efficacy; the therapeutic range for
pioglitazone is 15 to 45 mg/d in a single daily dose and for rosiglitazone, the
total daily dose is 2 to 8 mg/d-administered either once daily or twice daily
in divided doses. The ability of thiazolidinediones to influence other features
of the insulin resistance syndrome is under investigation.
receptor is found at highest levels in adipocytes but is expressed at lower
levels in many other insulin-sensitive tissues. Agonists of this receptor
promote adipocyte differentiation and may reduce insulin resistance in skeletal
muscle indirectly. Thiazolidinediones reduce the fasting plasma glucose by
improving peripheral glucose utilization and insulin sensitivity (Table
333-12). Circulating insulin levels decrease with use of the
thiazolidinediones, indicating a reduction in insulin resistance. Although
direct comparisons are not available, the two currently available
thiazolidinediones appear to have similar efficacy; the therapeutic range for
pioglitazone is 15 to 45 mg/d in a single daily dose and for rosiglitazone, the
total daily dose is 2 to 8 mg/d-administered either once daily or twice daily
in divided doses. The ability of thiazolidinediones to influence other features
of the insulin resistance syndrome is under investigation.
The
prototype of this class of drugs, thiazolidinediones, was withdrawn from the
U.S. market after reports of hepatotoxicity and an association with an
idiosyncratic liver reaction that sometimes led to hepatic failure. The two
other thiazolidinediones, rosiglitazone and pioglitazone, thus far do not appear
to induce the liver abnormalities seen with troglitazone. However, long-term
experience with the newer agents is limited. Consequently, the FDA recommends
measurement of liver function tests prior to initiating therapy with a
thiazolidinedione and at regular intervals (every two months for the first year
and then periodically). The thiazolidinediones raise LDL and HDL slightly and
lower triglycerides by 10 to 15%, but the clinical significance of these
changes is not known. Thiazolidinediones are associated with minor weight gain
(1 to 2 kg), a small reduction in the hematocrit, and a mild increase in plasma
volume. Cardiac function is not affected, but the incidence of peripheral edema
is increased. They are contraindicated in patients with liver disease or
congestive heart failure (class III or IV). Thiazolidinediones have been shown
to induce ovulation in premenopausal women with polycystic ovary syndrome (see
"Insulin Resistance Syndromes," above). Women should be warned about
the risk of pregnancy, since the safety of thiazolidinediones in pregnancy is
not established.
Insulin Therapy in Type 2 DM
Modest
doses of insulin are quite efficacious in controlling hyperglycemia in newly
diagnosed type 2 DM. Insulin should be considered as the initial therapy in
type 2 DM, particularly in lean individuals or those with severe weight loss,
in individuals with underlying renal or hepatic disease that precludes oral
glucose-lowering agents, or in individuals who are hospitalized or acutely ill.
Insulin therapy is ultimately required by a substantial number of individuals
with type 2 DM because of the progressive nature of the disorder and the
relative insulin deficiency that develops in patients with long-standing
diabetes.
Because
endogenous insulin secretion continues and is capable of providing some
coverage of mealtime caloric intake, insulin is usually initiated in a single
dose of intermediate-acting insulin (0.3 to 0.4 U/kg per day), given either
before breakfast or just before bedtime (or ultralente at bedtime). Since
fasting hyperglycemia and increased hepatic glucose production are prominent
features of type 2 DM, bedtime insulin is more effective in clinical trials
than a single dose of morning insulin. Some physicians prefer a relatively low,
fixed starting dose of intermediate-acting insulin (~15 to 20 units in the
morning and 5 to 10 units at bedtime) to avoid hypoglycemia. The insulin dose
may then be adjusted in 10% increments as dictated by SMBG results. Both
morning and bedtime intermediate insulin may be used in combination with oral
glucose-lowering agents (biguanides, ![]() -glucosidase
inhibitors, or thiazolidinediones).
-glucosidase
inhibitors, or thiazolidinediones).
Choice of Initial Glucose-Lowering Agent
Though
insulin is an effective primary therapy for type 2 DM, most patients and
physicians currently prefer oral glucose-lowering drugs as the initial
pharmacologic approach. The level of hyperglycemia should influence the initial
choice of therapy. Assuming maximal benefit of MNT and increased physical
activity has been realized, patients with mild to moderate hyperglycemia
[fasting plasma glucose <11.1 to 13.9 mmol/L (200 to 250 mg/dL)] often
respond well to a single oral glucose-lowering agent. Patients with more severe
hyperglycemia [fasting plasma glucose >13.9 mmol/L (250 mg/dL)] may respond
partially but are unlikely to achieve normoglycemia with oral monotherapy.
Nevertheless, many physicians prefer a stepwise approach that starts with a
single agent and adds a second agent to achieve the glycemic target (see
"Combination Therapy," below). Some physicians begin insulin in
individuals with severe hyperglycemia [fasting plasma glucose >13.9 to 16.7
mmol/L (250 to 300 mg/dL)]. This approach is based on the rationale that more
rapid glycemic control will reduce "glucose toxicity" to the islet
cells, improve endogenous insulin secretion, and possibly allow oral
glucose-lowering agents to be more effective. If this occurs, the insulin may
be discontinued.
Insulin
secretagogues, biguanides, ![]() -glucosidase
inhibitors, thiazolidinediones, and insulin are approved for monotherapy of
type 2 DM. Although each class of oral glucose-lowering agents has unique
advantages and disadvantages, certain generalizations apply: (1) insulin
secretagogues, biguanides, and thiazolidinediones improve glycemic control to a
similar degree (1 to 2% reduction in HbA1c) and are more effective than
-glucosidase
inhibitors, thiazolidinediones, and insulin are approved for monotherapy of
type 2 DM. Although each class of oral glucose-lowering agents has unique
advantages and disadvantages, certain generalizations apply: (1) insulin
secretagogues, biguanides, and thiazolidinediones improve glycemic control to a
similar degree (1 to 2% reduction in HbA1c) and are more effective than ![]() -glucosidase
inhibitors; (2) assuming a similar degree of glycemic improvement, no clinical
advantage to one class of drugs has been demonstrated, and any therapy that
improves glycemic control is beneficial; (3) insulin secretagogues and
-glucosidase
inhibitors; (2) assuming a similar degree of glycemic improvement, no clinical
advantage to one class of drugs has been demonstrated, and any therapy that
improves glycemic control is beneficial; (3) insulin secretagogues and ![]() -glucosidase
inhibitors begin to lower the plasma glucose immediately, whereas the
glucose-lowering effects of the biguanides and thiazolidinediones are delayed
by several weeks to months; (4) not all agents are effective in all individuals
with type 2 DM (primary failure); (5) biguanides,
-glucosidase
inhibitors begin to lower the plasma glucose immediately, whereas the
glucose-lowering effects of the biguanides and thiazolidinediones are delayed
by several weeks to months; (4) not all agents are effective in all individuals
with type 2 DM (primary failure); (5) biguanides, ![]() -glucosidase
inhibitors, and thiazolidinediones do not directly cause hypoglycemia; and (6)
most individuals will eventually require treatment with more than one class of
oral glucose-lowering agents, reflecting the progressive nature of type 2 DM.
-glucosidase
inhibitors, and thiazolidinediones do not directly cause hypoglycemia; and (6)
most individuals will eventually require treatment with more than one class of
oral glucose-lowering agents, reflecting the progressive nature of type 2 DM.
Considerable
clinical experience exists with sulfonylureas and metformin because they have
been available for several decades. It is assumed that the ![]() -glucosidase
inhibitors and thiazolidinediones, which are newer classes of oral
glucose-lowering drugs, will reduce DM-related complications by improving
glycemic control, although long-term data are not yet available. The
thiazolidinediones are theoretically attractive because they target a
fundamental abnormality in type 2 DM, namely insulin resistance. However, these
agents are currently more costly than others and require liver function
monitoring.
-glucosidase
inhibitors and thiazolidinediones, which are newer classes of oral
glucose-lowering drugs, will reduce DM-related complications by improving
glycemic control, although long-term data are not yet available. The
thiazolidinediones are theoretically attractive because they target a
fundamental abnormality in type 2 DM, namely insulin resistance. However, these
agents are currently more costly than others and require liver function
monitoring.
A
reasonable treatment algorithm for initial therapy proposes either a
sulfonylurea or metformin as initial therapy because of their efficacy, known
side-effect profile, and relatively low cost (Fig. 333-14). Metformin has the
advantage that it promotes mild weight loss, lowers insulin levels, improves
the lipid profile slightly, and may have a lower secondary failure rate.
However, there is no difference in response rate or degree of glycemic control
when metformin and sulfonylureas are compared in randomized, prospective
clinical trials. Based on SMBG results and the HbA1c, the dose of either the
sulfonylurea or metformin should be increased until the glycemic target is
achieved. ![]() -Glucosidase
inhibitors and thiazolidinediones are alternative, initial agents (Fig.
333-14).
-Glucosidase
inhibitors and thiazolidinediones are alternative, initial agents (Fig.
333-14).
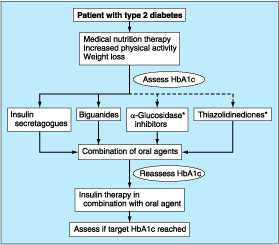
Figure 333-14: Glycemic management of
type 2 diabetes. See text for discussion. *See text about use as monotherapy.
The broken line indicates that biguanides or insulin secretagogues, but not ![]() glucosidase
inhibitors or thiazolidinediones, are preferred for initial therapy.
glucosidase
inhibitors or thiazolidinediones, are preferred for initial therapy.
When
used as monotherapy, approximately one-third of individuals will reach their target
glycemic goal with either a sulfonylurea or metformin. Approximately 25% of
individuals will not respond to sulfonylureas or metformin; under these
circumstances, the drug usually should be discontinued. Some individuals
respond to one agent but not the other. The remaining individuals treated with
either sulfonylureas or metformin alone will exhibit some improvement in
glycemic control but will not achieve their glycemic target and should be
considered for combination therapy.
Combination Therapy with Glucose-Lowering Agents
A
number of combinations of therapeutic agents are successful in type 2 DM, and
the dosing of agents in combination is the same as when the agents are used
alone. Because mechanisms of action of the first and second agents are different,
the effect on glycemic control is usually additive. Commonly used regimens
include: (1) insulin secretagogue with metformin or thiazolidinedione, (2)
sulfonylurea with ![]() -glucosidase
inhibitor, and (3) insulin with metformin or thiazolidinedione. The combination
of metformin and a thiazolidinedione is also effective and complementary. If
adequate control is not achieved with two oral agents, bedtime insulin or a
third oral agent may be added stepwise. However, long-term experience with any
triple combination is lacking, and experience with two-drug combinations is
relatively limited.
-glucosidase
inhibitor, and (3) insulin with metformin or thiazolidinedione. The combination
of metformin and a thiazolidinedione is also effective and complementary. If
adequate control is not achieved with two oral agents, bedtime insulin or a
third oral agent may be added stepwise. However, long-term experience with any
triple combination is lacking, and experience with two-drug combinations is
relatively limited.
Insulin
becomes required as type 2 DM enters the phase of relative insulin deficiency
(as seen in long-standing DM) and is signaled by inadequate glycemic control on
one or two oral glucose-lowering agents. Insulin can be used in combination
with any of the oral agents in patients who fail to reach the glycemic target.
For example, a single dose of intermediate-acting insulin at bedtime is
effective in combination with metformin. As endogenous insulin production falls
further, multiple injections of intermediate-acting and short-acting insulin
regimens are necessary to control postprandial glucose excursions. These
combination regimens are identical to the intermediate- and short-acting
combination regimens discussed above for type 1 DM. Since the hyperglycemia of
type 2 DM tends to be more "stable," these regimens can be increased
in 10% increments every 2 to 3 days using SMBG results. The daily insulin dose
required can become quite large (1 to 2 units/kg per day) as endogenous insulin
production falls and insulin resistance persists. Individuals who require >1
unit/kg per day of intermediate-acting insulin should be considered for
combination therapy with metformin or a thiazolidinedione. The addition of a
thiazolidinedione can reduce insulin requirements in some individuals with type
2 DM, while maintaining or even improving glycemic control.
Intensive
diabetes management (Table 333-10) is a treatment option in type 2 patients who
cannot achieve optimal glycemic control and are capable of implementing such
regimens. A recent study from the Veterans Administration found that intensive
diabetes management is not associated with a greater degree of side effects
(hypoglycemia, weight gain) than standard insulin therapy. The effect of higher
insulin levels associated with intensive diabetes management on the prognosis
of diseases commonly associated with type 2 DM (cardiovascular disease,
hypertension) is still debated. In selected patients with type 2 DM, insulin
pumps improve glycemic control and are well tolerated.
Emerging Therapies
Whole
pancreas transplantation (conventionally performed concomitantly with a renal
transplant) may normalize glucose tolerance and is an important therapeutic
option in type 1 diabetes, though it requires substantial expertise and is
associated with the side effects of immunosuppression. Pancreatic islet
transplantation has been plagued by limitations in pancreatic islet isolation
and graft survival, but recent advances in specific immunomodulation have
greatly improved the results. Islet transplantation is an area of active
clinical investigation.
Advances
in molecular biology and new insights into normal mechanisms of glucose
homeostasis have led to a number of emerging therapies for diabetes and its
complications. For example, glucagon-like peptide 1, a potent insulin
secretagogue, may be efficacious in type 2 DM. Inhaled insulin and additional
insulin analogues are in advanced stages of clinical trials. Aminoguanidine, an
inhibitor of the formation of advanced glycosylation end products, and
inhibitors of protein kinase C may reduce the complications of DM. Closed-loop
pumps that infuse the appropriate amount of insulin in response to changing
glucose levels are potentially feasible now that continuous glucose-monitoring
technology has been developed.
COMPLICATIONS OF THERAPY FOR DIABETES MELLITUS
As
with any therapy, the benefits of efforts directed towards glycemic control
must be weighed against the risks of treatment. Side effects of intensive
treatment include an increased frequency of serious hypoglycemia, weight gain,
increased economic costs, and greater demands on the patient. In the DCCT,
quality of life was very similar in the intensive therapy and standard therapy
groups. The most serious complication of therapy for DM is hypoglycemia (Chap.
334). Weight gain occurs with most (insulin, insulin secretagogues,
thiazolidinediones) but not all (metformin and ![]() -glucosidase
inhibitors) therapies that improve glycemic control due to the anabolic effects
of insulin and the reduction in glucosuria. In the DCCT, individuals with the
greatest weight gain exhibited increases in LDL cholesterol and triglycerides
as well as increases in blood pressure (both systolic and diastolic) similar to
those seen in individuals with type 2 DM and insulin resistance. These effects
could increase the risk of cardiovascular disease in intensively managed
patients. As discussed previously, improved glycemic control is sometimes
accompanied by a transient worsening of diabetic retinopathy or neuropathy.
-glucosidase
inhibitors) therapies that improve glycemic control due to the anabolic effects
of insulin and the reduction in glucosuria. In the DCCT, individuals with the
greatest weight gain exhibited increases in LDL cholesterol and triglycerides
as well as increases in blood pressure (both systolic and diastolic) similar to
those seen in individuals with type 2 DM and insulin resistance. These effects
could increase the risk of cardiovascular disease in intensively managed
patients. As discussed previously, improved glycemic control is sometimes
accompanied by a transient worsening of diabetic retinopathy or neuropathy.
ONGOING ASPECTS OF COMPREHENSIVE DIABETES CARE
The
morbidity and mortality of DM-related complications can be greatly reduced by
timely and consistent surveillance procedures (Table 333-14). These screening
procedures are indicated for all individuals with DM, but numerous studies have
documented that most individuals with diabetes do not receive comprehensive
diabetes care. Screening for dyslipidemia and hypertension should be performed
annually. In addition to routine health maintenance, individuals with diabetes
should also receive the pneumococcal and tetanus vaccines (at recommended
intervals) and the influenza vaccine (annually).
Table 333-14: Guidelines for Ongoing Medical
Care for Patients with Diabetes
|
An
annual comprehensive eye examination should be performed by a qualified
optometrist or ophthalmologist. If abnormalities are detected, further
evaluation and treatment require an ophthalmologist skilled in diabetes-related
eye disease. Because many individuals with type 2 DM have had asymptomatic
diabetes for several years before diagnosis, a consensus panel from the ADA
recommends the following ophthalmologic examination schedule: (1) individuals
with onset of DM at <29 years should have an initial eye examination within
3 to 5 years of diagnosis, (2) individuals with onset of DM at >30 years
should have an initial eye examination at the time of diabetes diagnosis, and
(3) women with DM who are contemplating pregnancy should have an eye
examination prior to conception and during the first trimester.
An
annual foot examination should: (1) assess blood flow, sensation, and nail
care; (2) look for the presence of foot deformities such as hammer or claw toes
and Charcot foot; and (3) identify sites of potential ulceration. Calluses and
nail deformities should be treated by a podiatrist; the patient should be
discouraged from self-care of even minor foot problems.
An
annual microalbuminuria measurement is advised in individuals with type 1 or
type 2 DM and no protein on a routine urinalysis (Fig. 333-10). If the
urinalysis detects proteinuria, the amount of protein should be quantified by
standard urine protein measurements. If the urinalysis was negative for protein
in the past, microalbuminuria should be the annual screening examination. Routine
urine protein measurements do not detect low levels of albumin excretion.
Screening should commence 5 years after the onset of type 1 DM and at the time
of onset of type 2 DM.
SPECIAL CONSIDERATIONS IN DIABETES MELLITUS
PSYCHOSOCIAL ASPECTS
As
with any chronic, debilitating disease, the individual with DM faces a series
of challenges that affect all aspects of daily life. The individual with DM
must accept that he or she may develop complications related to DM. Even with
considerable effort, normoglycemia can be an elusive goal, and solutions to
worsening glycemic control may not be easily identifiable. The patient should
view him- or herself as an essential member of the diabetes care team and not
as someone who is cared for by the diabetes team. Emotional stress may provoke
a change in behavior so that individuals no longer adhere to a dietary,
exercise, or therapeutic regimen. This can lead to the appearance of either
hyper- or hypoglycemia. Depression and eating disorders (in women) are more
common in individuals with type 1 or type 2 DM
MANAGEMENT IN THE HOSPITALIZED PATIENT
Virtually
all medical and surgical subspecialties may be involved in the care of
hospitalized patients with diabetes. General anesthesia, surgery, and concurrent
illness raise the levels of counterregulatory hormones (cortisol, growth
hormone, catecholamines, and glucagon), and infection may lead to transient
insulin resistance. These factors increase insulin requirements by increasing
glucose production and impairing glucose utilization and thus may worsen
glycemic control. On the other hand, the concurrent illness or surgical
procedure may prevent the patient with DM from eating normally and may promote
hypoglycemia. Glycemic control should be assessed (with HbA1c) and, if
feasible, should be optimized prior to surgery. Electrolytes, renal function,
and intravascular volume status should be assessed as well. The extremely high
prevalence of asymptomatic cardiovascular disease in individuals with DM
(especially in type 2 DM) may require preoperative cardiovascular evaluation.
The
goals of diabetes management during hospitalization are avoidance of
hypoglycemia, optimization of glycemic control, and transition back to the
outpatient diabetes treatment regimen. Attention to each stage in this process
requires integrating information regarding the plasma glucose, diabetes
treatment regimen, and clinical status of the patient. For example, some
surgical procedures utilizing local anesthesia or epidural anesthesia may have
minimal effects on glycemic control. If the patient is eating soon after the
procedure and there is no disruption of the patient's regular meal plans, then
glycemic control is usually maintained.
The
physician caring for an individual with diabetes in the perioperative period,
during times of infection or serious physical illness, or simply when fasting
for a diagnostic procedure must monitor the plasma glucose vigilantly, adjust
the diabetes treatment regimen, and provide glucose infusion as needed. Several
different treatment regimens (intravenous or subcutaneous insulin regimens) can
be employed successfully. Individuals with type 1 DM require continued insulin
administration to maintain the levels of circulating insulin necessary to
prevent DKA. Prolongation of a surgical procedure or delay in the recovery room
is not uncommon and may result in periods of insulin deficiency. Even
relatively brief periods without insulin may lead to mild DKA. Individuals with
type 1 DM who are undergoing general anesthesia and surgery, or who are
seriously ill, should receive continuous insulin, either through an intravenous
insulin infusion or by subcutaneous administration of a reduced dose of
long-acting insulin. Short-acting insulin alone is insufficient.
Individuals
with type 2 DM can be managed with either insulin infusion or a reduced dose of
subcutaneous insulin. Oral glucose-lowering agents are discontinued at the time
a combined insulin/glucose infusion is started. Oral agents such as
sulfonylureas, metformin, acarbose, and thiazolidinediones are not useful in
regulating the plasma glucose in clinical situations where the insulin
requirements and glucose intake are changing rapidly. Moreover, these oral
agents may be dangerous if the patient is fasting (e.g., hypoglycemia with
sulfonylureas). Metformin should be withheld when radiographic contrast media
will be given or if severe congestive heart failure, acidosis, or declining
renal function is present.
Insulin
infusions can effectively control plasma glucose in the perioperative period
and when the patient is unable to take anything by mouth. The absorption of
subcutaneous insulin may be variable in such situations because of changes in
blood flow. The physician must consider carefully the clinical setting in which
an insulin infusion will be utilized, including whether adequate ancillary
personnel are available to monitor the plasma glucose frequently and whether
they can adjust the insulin infusion rate, either based on an algorithm or in
consultation with the physician. The initial rate for an insulin infusion may
range from 0.5 to 5 units/h, depending on the degree of insulin resistance and
the clinical situation. Based on hourly capillary glucose measurements, the
insulin infusion rate is adjusted to maintain the plasma glucose within the
desired range [5.6 to 11.1 mmol/L (100 to 200 mg/dL)]. Glucose infusion,
initiated at the time the patient begins fasting, should be adjusted to deliver
the equivalent of 50 to 150 mL of D5W/h until the patient is
reliably taking nutrition orally. The insulin infusion can be temporarily
discontinued if hypoglycemia occurs and may be resumed at a lower infusion rate
once the plasma glucose exceeds 5.6 mmol/L (100 mg/dL).
Insulin
infusion is the preferred method for managing patients with type 1 DM in the
perioperative period or when serious concurrent illness is present. Individuals
with type 2 DM can be managed with an insulin infusion, but subcutaneous
insulin in reduced doses can be used effectively as well. If the diagnostic or
surgical procedure is brief and performed under local or regional anesthesia, a
reduced dose of subcutaneous, long-acting insulin may suffice. This approach
facilitates the transition back to the long-acting insulin after the procedure.
The dose of long-acting insulin should be reduced by 30 to 40%, and
short-acting insulin is either held or, likewise, reduced by 30 to 40%. Glucose
should be infused to prevent hypoglycemia.
Total Parenteral Nutrition
Total
parenteral nutrition (TPN) greatly increases insulin requirements. In addition,
individuals not previously known to have DM may become hyperglycemic during TPN
and require insulin treatment. Intravenous insulin infusion is the preferred
treatment for hyperglycemia, and rapid titration to the required insulin dose
is done most efficiently using a separate insulin infusion. After the total
insulin dose has been determined, insulin may be added directly to the TPN
solution. Often, individuals receiving either TPN or enteral nutrition receive
their caloric loads continuously and not at "meal times";
consequently, subcutaneous insulin regimens must be adjusted.
GLUCOCORTICOIDS
Glucocorticoids
increase insulin resistance, decrease glucose utilization, increase hepatic
glucose production, and impair insulin secretion. These changes lead to a
worsening of glycemic control in individuals with DM and may precipitate
diabetes in other individuals ("steroid-induced diabetes"). The
effects of glucocorticoids on glucose homeostasis are dose-related, usually
reversible, and most pronounced in the postprandial period. If the fasting
plasma glucose is near the normal range, oral diabetes agents (sulfonylureas
and acarbose) may be sufficient to reduce hyperglycemia. If the fasting plasma
glucose >11.1 mmol/L (200 mg/dL), oral agents are usually not efficacious
and insulin therapy is required. Short-acting insulin may be required to
supplement long-acting insulin in order to control postprandial glucose
excursions.
REPRODUCTIVE ISSUES
Reproductive
capacity in either men or women with DM appears to be normal. Menstrual cycles
may be associated with alterations in glycemic control in women with DM.
Pregnancy is associated with marked insulin resistance; the increased insulin
requirements often precipitate DM and lead to the diagnosis of GDM. Glucose,
which at high levels is a teratogen to the developing fetus, readily crosses
the placenta, but insulin does not. Thus, hyperglycemia or hypoglycemia from
the maternal circulation may stimulate insulin secretion in the fetus. The
anabolic and growth effects of insulin may result in macrosomia. GDM
complicates approximately 4% of pregnancies in the United States. The incidence
of GDM is greatly increased in certain ethnic groups, including African
Americans and Hispanic Americans, consistent with a similar increased risk of
type 2 DM. Current recommendations advise screening for glucose intolerance
between weeks 24 and 28 of pregnancy in women with high risk for GDM (![]() 25
years; obesity; family history of DM; member of an ethnic group such as
Hispanic American, Native American, Asian American, African American, or
Pacific Islander). Therapy for GDM is similar to that for individuals with
pregnancy-associated diabetes and involves MNT and insulin, if hyperglycemia
persists. Oral glucose-lowering agents have not been approved for use during
pregnancy. With current practices, the morbidity and mortality of the mother
with GDM and the fetus are no different from those in the nondiabetic
population. Individuals who develop GDM are at marked increased risk for
developing type 2 DM in the future and should be screened periodically for DM.
After delivery, glucose homeostasis should be reassessed in the mother. Most
individuals with GDM revert to normal glucose tolerance, but some will continue
to have overt diabetes or impairment of glucose tolerance. In addition,
children of women with GDM appear to be at risk for obesity and glucose
intolerance and have an increased risk of diabetes beginning in the later stages
of adolescence.
25
years; obesity; family history of DM; member of an ethnic group such as
Hispanic American, Native American, Asian American, African American, or
Pacific Islander). Therapy for GDM is similar to that for individuals with
pregnancy-associated diabetes and involves MNT and insulin, if hyperglycemia
persists. Oral glucose-lowering agents have not been approved for use during
pregnancy. With current practices, the morbidity and mortality of the mother
with GDM and the fetus are no different from those in the nondiabetic
population. Individuals who develop GDM are at marked increased risk for
developing type 2 DM in the future and should be screened periodically for DM.
After delivery, glucose homeostasis should be reassessed in the mother. Most
individuals with GDM revert to normal glucose tolerance, but some will continue
to have overt diabetes or impairment of glucose tolerance. In addition,
children of women with GDM appear to be at risk for obesity and glucose
intolerance and have an increased risk of diabetes beginning in the later stages
of adolescence.
Pregnancy
in individuals with known DM requires meticulous planning and adherence to
strict treatment regimens. Intensive diabetes management and normalization of
the HbA1c are the standard of care for individuals with existing DM who are
planning pregnancy. The crucial period of glycemic control is extremely early
following fertilization. The risk of fetal malformations is increased 4 to 10
times in individuals with uncontrolled DM at the time of conception. The goals
are normal plasma glucose during the preconception period and throughout the
periods of organ development in the fetus.
LIPODYSTROPHIC DM
Lipodystrophy,
or the loss of subcutaneous fat tissue, may be generalized in certain genetic
conditions such as leprechaunism. Generalized lipodystrophy is associated with
severe insulin resistance and is often accompanied by acanthosis nigricans and
dyslipidemia. Localized lipodystrophy associated with insulin injections has
been reduced considerably by the use of human insulin.
Protease Inhibitors and Lipodystrophy
Protease
inhibitors used in the treatment of HIV disease (Chap. 309) have been
associated with a centripetal accumulation of fat (visceral and abdominal
area), accumulation of fat in the dorsocervical region, loss of extremity fat,
decreased insulin sensitivity (elevations of the fasting insulin level and
reduced glucose tolerance on intravenous glucose tolerance testing), and
dyslipidemia. Although many aspects of the physical appearance of these
individuals resemble Cushing's syndrome, derangements in cortisol secretion
have not been found consistently and do not appear to account for this
appearance. Although some individuals have IGT, diabetes is not a common
feature. The possibility remains that this is related to HIV infection by some
undefined mechanism, since some features of the syndrome were observed before
the introduction of protease inhibitors. Therapy for HIV-related lipodystrophy
is not well established.