Jaundice
Jaundice,
or icterus, is a yellowish discoloration of tissue resulting from the
deposition of bilirubin. Tissue deposition of bilirubin occurs only in the
presence of serum hyperbilirubinemia and is a sign of either liver disease or,
less often, a hemolytic disorder. The degree of serum bilirubin elevation can
be estimated by physical examination. Slight increases in serum bilirubin are
best detected by examining the sclerae which have a particular affinity for
bilirubin due to their high elastin content. The presence of scleral icterus
indicates a serum bilirubin of at least 3.0 mg/dL. The ability to detect
scleral icterus is made more difficult if the examining room has fluorescent
lighting. If the examiner suspects scleral icterus, a second place to examine
is underneath the tongue. As serum bilirubin levels rise, the skin will
eventually become yellow in light-skinned patients and even green if the
process is longstanding; the green color is produced by oxidation of bilirubin
to biliverdin.
The
differential diagnosis for yellowing of the skin is limited. In addition to
jaundice, it includes carotenoderma, the use of the drug quinacrine, and
excessive exposure to phenols. Carotenoderma is the yellow color imparted to
the skin by the presence of carotene; it occurs in healthy individuals who
ingest excessive amounts of vegetables and fruits that contain carotene, such
as carrots, leafy vegetables, squash, peaches, and oranges. Unlike jaundice,
where the yellow coloration of the skin is uniformly distributed over the body,
in carotenoderma the pigment is concentrated on the palms, soles, forehead, and
nasolabial folds. Carotenoderma can be distinguished from jaundice by the
sparing of the sclerae. Quinacrine causes a yellow discoloration of the skin in
4 to 37% of patients treated with it. Unlike carotene, quinacrine can cause
discoloration of the sclerae.
Another
sensitive indicator of increased serum bilirubin is darkening of the urine,
which is due to the renal excretion of conjugated bilirubin. Patients often
describe their urine as tea or cola colored. Bilirubinuria indicates an
elevation of the direct serum bilirubin fraction and therefore the presence of
liver disease.
Increased
serum bilirubin levels occur when an imbalance exists between bilirubin
production and clearance. A logical evaluation of the patient who is jaundiced
requires an understanding of bilirubin production and metabolism.
Production and Metabolism of Bilirubin
Bilirubin,
a tetrapyrrole pigment, is a breakdown product of heme (ferroprotoporphyrin
IX). About 70 to 80% of the 250 to 300 mg of bilirubin produced each day is
derived from the breakdown of hemoglobin in senescent red blood cells. The
remainder comes from prematurely destroyed erythroid cells in bone marrow and
from the turnover of hemoproteins such as myoglobin and crytochromes found in
tissues throughout the body.
The
formation of bilirubin occurs in reticuloendothelial cells, primarily in the
spleen and liver. The first reaction, catalyzed by the enzyme heme oxygenase,
oxidatively cleaves the ![]() bridge
of the porphyrin group and opens the heme ring. The end products of this
reaction are biliverdin, carbon monoxide, and iron. The second reaction,
catalyzed by the cytosolic enzyme biliverdin reductase, reduces the central
methylene bridge of biliverdin and converts it to bilirubin. Bilirubin formed
in the reticuloendothelial cells is virtually insoluble in water. To be
transported in blood, it must be solubilized. This is accomplished by its
reversible, noncovalent binding to albumin. Unconjugated bilirubin bound to
albumin is transported to the liver, where it, but not the albumin, is taken up
by hepatocytes via a process that at least partly involves carrier-mediated
membrane transport.
bridge
of the porphyrin group and opens the heme ring. The end products of this
reaction are biliverdin, carbon monoxide, and iron. The second reaction,
catalyzed by the cytosolic enzyme biliverdin reductase, reduces the central
methylene bridge of biliverdin and converts it to bilirubin. Bilirubin formed
in the reticuloendothelial cells is virtually insoluble in water. To be
transported in blood, it must be solubilized. This is accomplished by its
reversible, noncovalent binding to albumin. Unconjugated bilirubin bound to
albumin is transported to the liver, where it, but not the albumin, is taken up
by hepatocytes via a process that at least partly involves carrier-mediated
membrane transport.
In
the cytosol of the hepatocyte, unconjugated bilirubin is coupled predominantly
to the protein ligandin (formerly called the Y protein). Ligandin was initially
thought to be a transport protein facilitating the movement of bilirubin from
the sinusodial membrane to the endoplasmic reticulum. It is now thought to slow
the cytosolic diffusion of bilirubin and to reduce its efflux back into serum.
In the endoplasmic reticulum, bilirubin is solubilized by conjugation to
glucuronic acid, forming bilirubin monoglucuronide and diglucuronide. The
conjugation of glucuronic acid to bilirubin is catalyzed by bilirubin
uridine-diphosphate (UDP) glucuronosyltransferase.
The
now hydrophilic bilirubin conjugates diffuse from the endoplasmic reticulum to
the canalicular membrane, where bilirubin monoglucuronide and diglucuronide are
actively transported into canalicular bile by an energy-dependent mechanism
involving the multiple organic ion transport protein/multiple drug resistance
protein. The conjugated bilirubin excreted into bile drains into the duodenum
and passes unchanged through the proximal small bowel. Conjugated bilirubin is
not taken up by the intestinal mucosa. When the conjugated bilirubin reaches
the distal ileum and colon, it is hydrolyzed to unconjugated bilirubin by
bacterial ![]() -glucuronidases.
The unconjugated bilirubin is reduced by normal gut bacteria to form a group of
colorless tetrapyrroles called urobilinogens. About 80 to 90% of these products
are excreted in feces, either unchanged or oxidized to orange derivatives
called urobilins. The remaining 10 to 20% of the urobilinogens are passively
absorbed, enter the portal venous blood, and are reexcreted by the liver. A
small fraction (usually less than 3 mg/dL) escapes hepatic uptake, filters
across the renal glomerulus, and is excreted in urine.
-glucuronidases.
The unconjugated bilirubin is reduced by normal gut bacteria to form a group of
colorless tetrapyrroles called urobilinogens. About 80 to 90% of these products
are excreted in feces, either unchanged or oxidized to orange derivatives
called urobilins. The remaining 10 to 20% of the urobilinogens are passively
absorbed, enter the portal venous blood, and are reexcreted by the liver. A
small fraction (usually less than 3 mg/dL) escapes hepatic uptake, filters
across the renal glomerulus, and is excreted in urine.
Measurement of Serum Bilirubin
The
terms direct- and indirect-reacting bilirubin are based on the original van den
Bergh reaction. This assay, or a variation of it, is still used in most
clinical chemistry laboratories to determine the serum bilirubin level. In this
assay, bilirubin is exposed to diazotized sulfanilic acid, splitting into two
relatively stable dipyrrylmethene azopigments that absorb maximally at 540 nm,
allowing for photometric analysis. The direct fraction is that which reacts
with diazotized sulfanilic acid in the absence of an accelerator substance such
as alcohol. The direct fraction provides an approximate determination of the
conjugated bilirubin in serum. The total serum bilirubin is the amount that reacts
after the addition of alcohol. The indirect fraction is the difference between
the total and the direct bilirubin and provides an estimate of the unconjugated
bilirubin in serum.
With
the van den Bergh method, the normal serum bilirubin concentration usually is
<1 mg/dL (17 ![]() mol/L).
Up to 30%, or 0.3 mg/dL (5.1
mol/L).
Up to 30%, or 0.3 mg/dL (5.1 ![]() mol/L),
of the total may be direct-reacting (conjugated) bilirubin. Total serum
bilirubin concentrations are between 0.2 and 0.9 mg/dL in 95% of a normal
population.
mol/L),
of the total may be direct-reacting (conjugated) bilirubin. Total serum
bilirubin concentrations are between 0.2 and 0.9 mg/dL in 95% of a normal
population.
Several
new techniques, although less convenient to perform, have added considerably to
our understanding of bilirubin metabolism. First, they demonstrate that in
normal people or those with Gilbert's syndrome, almost 100% of the serum
bilirubin is unconjugated; less than 3% is monoconjugated bilirubin. Second, in
jaundiced patients with hepatobiliary disease, the total serum bilirubin
concentration measured by these new, more accurate methods is lower than the
values found with diazo methods. This suggests that there are diazo-positive
compounds distinct from bilirubin in the serum of patients with hepatobiliary
disease. Third, these studies indicate that in jaundiced patients with
hepatobiliary disease, monoglucuronides of bilirubin predominate over the
diglucuronides. Fourth, part of the direct-reacting bilirubin fraction includes
conjugated bilirubin that is covalently linked to albumin. This albumin-linked
bilirubin fraction (delta fraction or biliprotein) represents an
important fraction of total serum bilirubin in patients with cholestasis and
hepatobiliary disorders. Albumin-bound conjugated bilirubin is formed in serum
when hepatic excretion of bilirubin glucuronides is impaired and the
glucuronides are present in serum in increasing amounts. By virtue of its tight
binding to albumin, the clearance rate of albumin-bound bilirubin from serum
approximates the half-life of albumin, 12 to 14 days, rather than the short
half-life of bilirubin, about 4 h.
The
prolonged half-life of albumin-bound conjugated bilirubin explains two
previously unexplained enigmas in jaundiced patients with liver disease: (1)
that some patients with conjugated hyperbilirubinemia do not exhibit
bilirubinuria during the recovery phase of their disease because the bilirubin
is bound to albumin and therefore not filtered by the renal glomeruli and (2)
that the elevated serum bilirubin level declines more slowly than expected in
some patients who otherwise appear to be recovering satisfactorily. Late in the
recovery phase of hepatobiliary disorders, all the conjugated bilirubin may be
in the albumin-linked form. Its value in serum falls slowly because of the long
half-life of albumin.
Measurement of Urine Bilirubin
Unconjugated
bilirubin is always bound to albumin in the serum, is not filtered by the
kidney, and is not found in the urine. Conjugated bilirubin is filtered at the
glomerulus and the majority is reabsorbed by the proximal tubules; a small
fraction is excreted in the urine. Any bilirubin found in the urine is
conjugated bilirubin. The presence of bilirubinuria implies the presence of
liver disease. A urine dipstick test (Ictotest) gives the same information as
fractionation of the serum bilirubin. This test is very accurate. A
false-negative test is possible in patients with prolonged cholestasis due to
the predominance of conjugated bilirubin covalently bound to albumin.
The Evaluation of Jaundice
The
bilirubin present in serum represents a balance between input from production
of bilirubin and hepatic/biliary removal of the pigment. Hyperbilirubinemia may
result from (1) overproduction of bilirubin; (2) impaired uptake, conjugation,
or excretion of bilirubin; or (3) regurgitation of unconjugated or conjugated
bilirubin from damaged hepatocytes or bile ducts. An increase in unconjugated bilirubin
in serum results from either overproduction, impairment of uptake, or
conjugation of bilirubin. An increase in conjugated bilirubin is due to
decreased excretion into the bile ductules or backward leakage of the pigment.
The initial steps in evaluating the patient with jaundice are to determine (1)
whether the hyperbilirubinemia is predominantly conjugated or unconjugated in
nature, and (2) whether other biochemical liver tests are abnormal. The
thoughtful interpretation of limited data will allow for a rational evaluation
of the patient (Fig. 45-1). This discussion will focus solely on the evaluation
of the adult patient with jaundice.
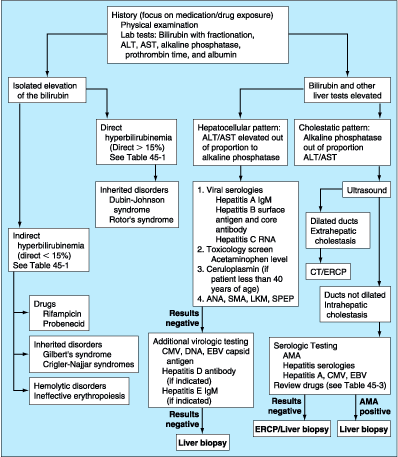
Figure 45-1: Evaluation of the patient with jaundice.
ERCP, endoscopic retrograde cholangiopancreatography; CT, computed tomography;
ALT, alanine aminotransferase; AST, aspartate aminotransferase; SMA, smooth
muscle antibody; AMA, antimitochondrial antibody; LKM, liver-kidney microsomal
antibody; SPEP, serum protein electrophoresis; CMV, cytomegalovirus; EBV,
Epstein-Barr virus.
Isolated Elevation of Serum Bilirubin
Unconjugated Hyperbilirubinemia
The
differential diagnosis of an isolated unconjugated hyperbilirubinemia is
limited (Table 45-1). The critical determination is whether the patient is
suffering from a hemolytic process resulting in an overproduction of bilirubin
(hemolytic disorders and ineffective erythropoiesis) or from impaired hepatic
uptake/conjugation of bilirubin (drug effect or genetic disorders).
Table 45-1: Causes of Isolated
Hyperbilirubinemia
|
Hemolytic
disorders that cause excessive heme production may be either inherited or acquired.
Inherited disorders include spherocytosis, sickle cell anemia, and deficiency
of red cell enzymes such as pyruvate kinase and glucose-6-phosphate
dehydrogenase. In these conditions, the serum bilirubin rarely exceeds 5 mg/dL.
Higher levels may occur when there is coexistent renal or hepatocellular
dysfunction, or in acute hemolysis such as a sickle cell crisis. In evaluating
jaundice in patients with chronic hemolysis, it is important to remember the
high incidence of pigmented (calcium bilirubinate) gallstones found in these
patients, which increases the likelihood of choledocholithiasis as an
alternative explanation for hyperbilirubinemia.
Acquired
hemolytic disorders include microangiopathic hemolytic anemia (e.g.,
hemolytic-uremic syndrome), paroxysmal nocturnal hemoglobinuria, and immune
hemolysis. Ineffective erythropoiesis occurs in cobalamin, folate, and iron
deficiencies.
In
the absence of hemolysis, the physician should consider a problem with the
hepatic uptake or conjugation of bilirubin. Certain drugs, including rifampicin
and probenecid, may cause unconjugated hyperbilirubinemia by diminishing
hepatic uptake of bilirubin. Impaired bilirubin conjugation occurs in three
genetic conditions: Crigler-Najjar syndrome, types I and II, and Gilbert's
syndrome. Crigler-Najjar type I is an exceptionally rare condition
found in neonates and characterized by severe jaundice (bilirubin > 20
mg/dL) and neurologic impairment due to kernicterus, frequently leading to
death in infancy or childhood. These patients have a complete absence of
bilirubin UDP glucuronosyltransferase activity, usually due to mutations in the
critical 3′ domain of the UDP glucuronosyltransferase gene, and are
totally unable to conjugate, hence cannot excrete bilirubin. The only effective
treatment is orthotopic liver transplantation. Use of gene therapy and
allogeneic hepatocyte infusion are experimental approaches of future promise
for this devastating disease.
Crigler-Najjar type II is somewhat more common than type I.
Patients live into adulthood with serum bilirubin levels that range from 6 to
25 mg/dL. In these patients, mutations in the bilirubin UDP
glucuronosyltransferase gene cause reduced but not completely absent activity
of the enzyme. Bilirubin UDP glucuronosyltransferase activity can be induced by
the administration of phenobarbital, which can reduce serum bilirubin levels in
these patients. Despite marked jaundice, these patients usually survive into
adulthood, although they may be susceptible to kernicterus under the stress of
intercurrent illness or surgery.
Gilbert's syndrome is also marked by the impaired conjugation of
bilirubin due to reduced bilirubin UDP glucuronosyltransferase activity.
Molecular analyses show that Gilbert's syndrome is due to reduced expression of
UDP glucuronosyltransferase activity caused by lengthening of the TATAA box
from A(TA)6 TAA to A(TA)7 TAA in the promoter element of
the gene. This results in mild unconjugated hyperbilirubinemia with serum
levels almost always less than 6 mg/dL. The serum levels may fluctuate and
jaundice is often identified only during periods of fasting. Unlike both
Crigler-Najjar syndromes, Gilbert's syndrome is very common. The reported
incidence is 3 to 7% of the population with males predominating over females by
a ratio of 2-7:1.
Conjugated Hyperbilirubinemia
Elevated
conjugated hyperbilirubinemia is found in two rare inherited conditions: Dubin-Johnson
syndrome and Rotor's syndrome (Table 45-1). Patients with both
conditions present with asymptomatic jaundice, typically in the second
generation of life. The defect in Dubin-Johnson syndrome is a point mutation in
the gene for the canalicular multispecific organic anion transporter. These
patients have altered excretion of bilirubin into the bile ducts. Rotor's syndrome
seems to be a problem with the hepatic storage of bilirubin. Differentiating
between these syndromes is possible, but clinically unnecessary, due to their
benign nature.
Elevation of Serum Bilirubin with Other Liver
Test Abnormalities
The
remainder of this chapter will focus on the evaluation of the patient with a
conjugated hyperbilirubinemia in the setting of other liver test abnormalities.
This group of patients can be divided into those with a primary hepatocellular
process and those with intra- or extrahepatic cholestasis. Being able to make
this differentiation will guide the physician's evaluation (Fig. 45-1). This
differentiation is made on the basis of the history and physical examination as
well as the pattern of liver test abnormalities.
History
A
complete medical history is perhaps the single most important part of the
evaluation of the patient with unexplained jaundice. Important considerations
include the use of or exposure to any chemical or medication, either
physician-prescribed or over-the-counter, such as herbal and vitamin
preparations and other drugs such as anabolic steroids. The patient should be
carefully questioned about possible parenteral exposures, including
transfusions, intravenous and intranasal drug use, tattoos, and sexual activity.
Other important questions include recent travel history, exposure to people
with jaundice, exposure to possibly contaminated foods, occupational exposure
to hepatotoxins, alcohol consumption, the duration of jaundice, and the
presence of any accompanying symptoms such as arthralgias, myalgias, rash,
anorexia, weight loss, abdominal pain, fever, pruritis, and changes in the
urine and stool. While none of these latter symptoms are specific for any one
condition, they can suggest a particular diagnosis. A history of arthralgias
and myalgias predating jaundice suggests hepatitis, either viral or
drug-related. Jaundice associated with the sudden onset of severe right upper
quadrant pain and shaking chills suggests choledocholithiasis and ascending
cholangitis.
Physical Examination
The
general assessment should include assessment of the patient's nutritional
status. Temporal and proximal muscle wasting suggests longstanding diseases
such as pancreatic cancer or cirrhosis. Stigmata of chronic liver disease,
including spider nevi, palmar erythema, gynecomastia, caput medusae,
Dupuytren's contractures, parotid gland enlargement, and testicular atrophy are
commonly seen in advanced alcoholic (Laennec's) cirrhosis and occasionally in
other types of cirrhosis. An enlarged left supraclavicular node (Virchow's
node) or periumbilical nodule (Sister Mary Joseph's nodule) suggest an
abdominal malignancy. Jugular venous distention, a sign of right-sided heart
failure, suggests hepatic congestion. Right pleural effusion, in the absence of
clinically apparent ascites, may be seen in advanced cirrhosis.
The
abdominal examination should focus on the size and consistency of the liver,
whether the spleen is palpable and hence enlarged, and whether there is ascites
present. Patients with cirrhosis may have an enlarged left lobe of the liver
which is felt below the xiphoid and an enlarged spleen. A grossly enlarged
nodular liver or an obvious abdominal mass suggests malignancy. An enlarged
tender liver could be viral or alcoholic hepatitis or, less often, an acutely
congested liver secondary to right-sided heart failure. Severe right upper
quadrant tenderness with respiratory arrest on inspiration (Murphy's sign)
suggests cholecystitis or, occasionally, ascending cholangitis. Ascites in the
presence of jaundice suggests either cirrhosis or malignancy with peritoneal
spread.
Laboratory Tests
When
the physician encounters a patient with unexplained jaundice, there are a
battery of tests that are helpful in the initial evaluation. These include
total and direct serum bilirubin with fractionation, aminotransferases,
alkaline phosphatase, albumin, and prothrombin time tests. Enzyme tests
[alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline
phosphatase] are helpful in differentiating between a hepatocellular process
and a cholestatic process (see Table 293-1 and Fig. 45-1), a critical step in
determining what additional workup is indicated. Patients with a hepatocellular
process generally have a disproportionate rise in the aminotransferases
compared to the alkaline phosphatase. Patients with a cholestatic process have
a disproportionate rise in the alkaline phosphatase compared to the
aminotransferases. The bilirubin can be prominently elevated in both hepatocellular
and cholestatic conditions and therefore is not necessarily helpful in
differentiating between the two.
Table 293-1: Liver Test Patterns in
Hepatobiliary Disorders
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In
addition to the enzyme tests, all jaundiced patients should have additional
blood tests, specifically an albumin and a prothrombin time, to assess liver
function. A low albumin suggests a chronic process such as cirrhosis or cancer.
A normal albumin is suggestive of a more acute process such as viral hepatitis
or choledocholithiasis. An elevated prothrombin time indicates either vitamin K
deficiency due to prolonged jaundice and malabsorption of vitamin K or
significant hepatocellular dysfunction. The failure of the prothrombin time to
correct with parenteral administration of vitamin K indicates severe
hepatocellular injury.
The
results of the bilirubin, enzyme, albumin, and prothrombin time tests will
usually indicate whether a jaundiced patient has a hepatocellular or a
cholestatic disease. The causes and evaluation of each of these is quite
different.
Hepatocellular Conditions
Hepatocellular
diseases that can cause jaundice include viral hepatitis, drug or environmental
toxicity, alcohol, and end-stage cirrhosis from any cause (Table 45-2).
Wilson's disease should be considered in young adults. Autoimmune hepatitis is
typically seen in young to middle-aged women, but may affect men and women of
any age. Alcoholic hepatitis can be differentiated from viral and toxin-related
hepatitis by the pattern of the aminotransferases. Patients with alcoholic
hepatitis typically have an AST:ALT ratio of at least 2:1. The AST rarely
exceeds 300 U/L. Patients with acute viral hepatitis and toxin-related injury
severe enough to produce jaundice typically have aminotransferases greater than
500 U/L, with the ALT greater than or equal to the AST. The degree of
aminotransferase elevation can occasionally help in differentiating between
hepatocellular and cholestatic processes. While ALT and AST values less than 8
times normal may be seen in either hepatocellular or cholestatic liver disease,
values 25 times normal or higher are seen primarily in acute hepatocellular
diseases. Patients with jaundice from cirrhosis can have normal or only slight
elevations of the aminotransferases.
Table 45-2: Hepatocellular Conditions That May
Produce Jaundice
|
When
the physician determines that the patient has a hepatocellular disease,
appropriate testing for acute viral hepatitis includes a hepatitis A IgM
antibody, a hepatitis B surface antigen and core IgM antibody, and a hepatitis
C viral RNA test. It can take many weeks for the hepatitis C antibody to become
detectable, making it an unreliable test if acute hepatitis C is suspected.
Depending on circumstances, studies for hepatitis D, E, Epstein-Barr virus
(EBV), and cytomegalovirus (CMV) may be indicated. Ceruloplasmin is the initial
screening test for Wilson's disease. Testing for autoimmune hepatitis usually
includes an antinuclear antibody and measurement of specific immunoglobulins.
Drug-induced
hepatocellular injury can be classified either as predictable or unpredictable.
Predictable drug reactions are dose-dependent and affect all patients who
ingest a toxic dose of the drug in question. The classic example is
acetaminophen hepatotoxicity. Unpredictable or idiosyncratic drug reactions are
not dose-dependent and occur in a minority of patients. A great number of drugs
can cause idiosyncratic hepatic injury. Environmental toxins are also an
important cause of hepatocellular injury. Examples include industrial chemicals
such as vinyl chloride, herbal preparations containing pyrrolizidine alkaloids
(Jamaica bush tea), and the mushrooms Amanita phalloides or verna containing
highly hepatotoxic amatoxins.
Cholestatic Conditions
When
the pattern of the liver tests suggests a cholestatic disorder, the next step
is to determine whether it is intra- or extrahepatic cholestasis (Fig. 45-1).
Distinguishing intrahepatic from extrahepatic cholestasis may be difficult.
History, physical examination, and laboratory tests are often not helpful. The
next appropriate test is an ultrasound. The ultrasound is inexpensive, does not
expose the patient to ionizing radiation, and can detect dilation of the intra-
and extrahepatic biliary tree with a high degree of sensitivity and
specificity. The absence of biliary dilatation suggests intrahepatic
cholestasis, while the presence of biliary dilatation indicates extrahepatic
cholestasis. False-negative results occur in patients with partial obstruction
of the common bile duct or in patients with cirrhosis or primary sclerosing
cholangitis (PSC) where scarring prevents the intrahepatic ducts from dilating.
Although
ultrasonography may indicate extrahepatic cholestasis, it rarely identifies the
site or cause of obstruction. The distal common bile duct is a particularly
difficult area to visualize by ultrasound because of overlying bowel gas.
Appropriate next tests include computed tomography (CT) and endoscopic
retrograde cholangiopancreatography (ERCP). CT scanning is better than
ultrasonography for assessing the head of the pancreas and for identifying
choledocholithiasis in the distal common bile duct, particularly when the ducts
are not dilated. ERCP is the gold standard for identifying choledocholithiasis.
It is performed by introducing a side-viewing endoscope perorally into the
duodenum. The ampulla of Vater is visualized and a catheter is advanced through
the ampulla. Injection of dye allows for the visualization of the common bile
duct and the pancreatic duct. The success rate for cannulation of the common
bile duct ranges from 80 to 95%, depending on the operator's experience. Beyond
its diagnostic capabilities, ERCP allows for therapeutic interventions,
including the removal of common bile duct stones and the placement of stents.
In patients in whom ERCP is unsuccessful, transhepatic cholangiography can
provide the same information. Magnetic resonance cholangiopancreatography
(MRCP) is a rapidly developing, noninvasive technique for imaging the bile and
pancreatic ducts; this may replace ERCP as the initial diagnostic test in cases
where the need for intervention is felt to be small.
In
patients with apparent intrahepatic cholestasis, the diagnosis is often
made by serologic testing in combination with percutaneous liver biopsy. The
list of possible causes of intrahepatic cholestasis is long and varied (Table
45-3). A number of conditions that typically cause a hepatocellular pattern of
injury can also present as a cholestatic variant. Both hepatitis B and C can
cause a cholestatic hepatitis (fibrosing cholestatic hepatitis) that has
histologic features that mimic large duct obstruction. This disease variant has
been reported in patients who have undergone solid organ transplantation.
Hepatitis A, alcoholic hepatitis, EBV, and CMV may also present as cholestatic
liver disease.
Table 45-3: Cholestatic Conditions That May
Produce Jaundice
|
Drugs
may cause intrahepatic cholestasis, a variant of drug-induced hepatitis.
Drug-induced cholestasis is usually reversible after eliminating the offending
drug, although it may take many months for cholestasis to resolve. Drugs most
commonly associated with cholestasis are the anabolic and contraceptive
steroids. Cholestatic hepatitis has been reported with chlorpromazine,
imipramine, tolbutamide, sulindac, cimetidine, and erythromycin estolate. It
also occurs in patients taking trimethoprim, sulfamethoxazole, and
penicillin-based antibiotics such as ampicillin, dicloxacillin, and clavulinic
acid. Rarely, cholestasis may be chronic and associated with progressive
fibrosis despite early discontinuation of the drug. Chronic cholestasis has
been associated with chlorpromazine and prochlorperazine.
Primary biliary cirrhosis is a disease predominantly of
middle-aged women in which there is a progressive destruction of interlobular
bile ducts. The diagnosis is made by the presence of the antimitochondrial
antibody that is found in 95% of patients. Primary sclerosing cholangitis
(PSC) is characterized by the destruction and fibrosis of larger bile ducts.
The disease may involve only the intrahepatic ducts and present as intrahepatic
cholestasis. However, in 65% of patients with PSC, both intra- and extrahepatic
ducts are involved. The diagnosis of PSC is made by ERCP. The pathognomonic
findings are multiple strictures of bile ducts with dilatations proximal to the
strictures. Approximately 75% of patients with PSC have inflammatory bowel
disease.
The
vanishing bile duct syndrome and adult bile ductopenia are rare
conditions in which there are a decreased number of bile ducts seen in liver
biopsy specimens. The histologic picture is similar to that found in primary biliary
cirrhosis. This picture is seen in patients who develop chronic rejection after
liver transplantation and in those who develop graft-versus-host disease after
bone marrow transplantation. Vanishing bile duct syndrome also occurs in rare
cases of sarcoidosis, in patients taking certain drugs including
chlorpromazine, and idiopathically. There are also familial forms of
intrahepatic cholestasis, including the familial intrahepatic cholestatic
syndromes, I-III. Benign recurrent cholestasis is an autosomal recessive
disease that appears to be due to mutations in a P type ATPase, which probably
acts as a bile acid transporter. The disease is marked by recurrent episodes of
jaundice and pruritis; the episodes are self-limited but can be debilitating. Cholestasis
of pregnancy occurs in the second and third trimesters and resolves after
delivery. Its cause is unknown, but the condition is probably inherited and
cholestasis can be triggered by estrogen administration.
Other
causes of intrahepatic cholestasis include total parenteral nutrition (TPN),
nonhepatobiliary sepsis, benign postoperative cholestasis, and a paraneoplastic
syndrome associated with a number of different malignancies, including
Hodgkin's disease, medullary thyroid cancer, hypernephroma, renal sarcoma, T
cell lymphoma, prostate cancer, and several GI malignancies. In patients
developing cholestasis in the intensive care unit, the major considerations
should be sepsis, shock liver, and TPN jaundice. Jaundice occurring after bone
marrow transplantation is most likely due to venoocclusive disease or
graft-versus-host disease.
Causes
of extrahepatic cholestasis can be split into malignant and benign
(Table 45-3). Malignant causes include pancreatic, gallbladder, ampullary, and
cholangiocarcinoma. The latter is most commonly associated with PSC and is
exceptionally difficult to diagnose because its appearance is often identical
to PSC. Pancreatic and gallbladder tumors, as well as cholangiocarcinoma, are
rarely resectable and have poor prognoses. Ampullary carcinoma has the highest
surgical cure rate of all the tumors that present as painless jaundice. Hilar
lymphadenopathy due to metastases from other cancers may cause obstruction of
the extrahepatic biliary tree.
Choledocholithiasis is the most common cause of extrahepatic
cholestasis. The clinical presentation can range from mild right upper quadrant
discomfort with only minimal elevations of the enzyme tests to ascending
cholangitis with jaundice, sepsis, and circulatory collapse. PSC may occur with
clinically important strictures limited to the extrahepatic biliary tree. In
cases where there is a dominant stricture, patients can be effectively managed
with serial endoscopic dilatations. Chronic pancreatitis rarely causes
strictures of the distal common bile duct, where it passes through the head of
the pancreas. AIDS cholangiopathy is a condition, usually due to infection of
the bile duct epithelium with CMV or cryptosporidium, which has a
cholangiographic appearance similar to PSC. These patients usually present with
greatly elevated serum alkaline phosphatase levels, mean of 800 IU/L, but the
bilirubin is often near normal. These patients do not typically present with
jaundice.
Summary
The
goal of this chapter is not to provide an encyclopedic review of all of the
conditions that can cause jaundice. Rather, it is intended to provide a
framework that helps a physician to evaluate the patient with jaundice in a
logical way (Fig. 45-1).
Simply
stated, the initial step is to obtain appropriate blood tests to determine if
the patient has an isolated elevation of serum bilirubin. If so, is the
bilirubin elevation due to an increased unconjugated or conjugated fraction? If
the hyperbilirubinemia is accompanied by other liver test abnormalities, is the
disorder hepatocellular or cholestatic? If cholestatic, is it intra- or
extrahepatic? All of these questions can be answered with a thoughtful history,
physical examination, and interpretation of laboratory and radiologic tests and
procedures.