Acute Myocardial Infarction
Acute
myocardial infarction (AMI) is one of the most common diagnoses in hospitalized
patients in industrialized countries. In the United States, approximately 1.1
million AMIs occur each year. The mortality rate with AMI is approximately 30%,
with more than half of these deaths occurring before the stricken individual
reaches the hospital. Although the mortality rate after admission for AMI has
declined by about 30% over the last two decades, approximately 1 of every 25
patients who survives the initial hospitalization dies in the first year after
AMI. Survival is markedly reduced in elderly patients (over age 75), whose
mortality rate is 20% at 1 month and 30% at 1 year after AMI.
Pathophysiology:
Role of Acute Plaque Rupture
AMI
generally occurs when coronary blood flow decreases abruptly after a thrombotic
occlusion of a coronary artery previously narrowed by atherosclerosis. Slowly
developing, high-grade coronary artery stenoses usually do not precipitate AMI
because of the development of a rich collateral network over time. Instead, AMI
occurs when a coronary artery thrombus develops rapidly at a site of vascular
injury. This injury is produced or facilitated by factors such as cigarette
smoking, hypertension, and lipid accumulation. In most cases, infarction occurs
when an atherosclerotic plaque fissures, ruptures, or ulcerates and when
conditions (local or systemic) favor thrombogenesis, so that a mural thrombus
forms at the site of rupture and leads to coronary artery occlusion (Fig.
243-5). Histologic studies indicate that the coronary plaques prone to rupture
are those with a rich lipid core and a thin fibrous cap (Chap. 241). After an
initial platelet monolayer forms at the site of the ruptured plaque, various
agonists (collagen, ADP, epinephrine, serotonin) promote platelet activation.
After agonist stimulation of platelets, there is production and release of
thromboxane A2 (a potent local vasoconstrictor), further platelet
activation, and potential resistance to thrombolysis.
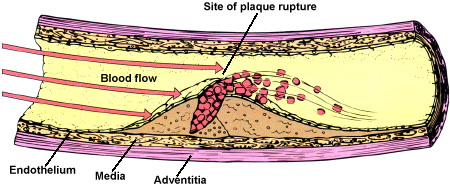
Figure 243-5: Diagram of arterial thrombus responsible
for acute myocardial infarction. Platelet adhesion and aggregation occur at the
site of plaque rupture. Activated platelets exert procoagulant effects and the
soluble coagulation cascade is activated. Fibrin strands and erythrocytes
predominate within the lumen of the vessel and downstream in the 'body' and
'tail' of the thrombus.[From NS Kleiman: Antiplatelet therapy, in RM Califf
(volume ed): Acute Myocardial Infarction and Other Acute Ischemic Syndromes, in
E Braunwald (series ed): Atlas of Heart Diseases, Philadelphia: Current
Medicine, 1996, p. 8.2.]
In
addition to the generation of thromboxane A2, activation of
platelets by agonists promotes a conformational change in the glycoprotein
IIb/IIIa receptor (Chap. 116). Once converted to its functional state, this
receptor develops a high affinity for amino acid sequences on soluble adhesive
proteins (i.e., integrins) such as von Willebrand factor (vWF) and fibrinogen.
Since vWF and fibrinogen are multivalent molecules, they can bind to two
different platelets simultaneously, resulting in platelet cross-linking and
aggregation.
The
coagulation cascade is activated on exposure of tissue factor in damaged
endothelial cells at the site of the ruptured plaque. Factors VII and X are
activated, ultimately leading to the conversion of prothrombin to thrombin,
which then converts fibrinogen to fibrin (Chap. 117). Fluid-phase and
clot-bound thrombin participate in an autoamplification reaction that leads to
further activation of the coagulation cascade. The culprit coronary artery
eventually becomes occluded by a thrombus containing platelet aggregates and
fibrin strands.
In
rare cases, AMI may be due to coronary artery occlusion caused by coronary
emboli, congenital abnormalities, coronary spasm, and a wide variety of
systemic-particularly inflammatory-diseases. The amount of myocardial damage
caused by coronary occlusion depends on (1) the territory supplied by the
affected vessel, (2) whether or not the vessel becomes totally occluded, (3)
the duration of coronary occlusion, (4) the quantity of blood supplied by
collateral vessels to the affected tissue, (5) the demand for oxygen of the
myocardium whose blood supply has been suddenly limited, (6) native factors
that can produce early spontaneous lysis of the occlusive thrombus, and (7) the
adequacy of myocardial perfusion in the infarct zone when flow is restored in
the occluded epicardial coronary artery.
Patients
at increased risk of developing AMI include those with multiple coronary risk
factors (Chap. 241) and those with unstable angina or Prinzmetal's variant
angina (Chap. 244). Less common underlying medical conditions predisposing
patients to AMI include hypercoagulability, collagen vascular disease, cocaine
abuse, and intracardiac thrombi or masses that can produce coronary emboli.
Clinical Presentation
In
up to one-half of cases, a precipitating factor appears to be present before
AMI, such as vigorous physical exercise, emotional stress, or a medical or
surgical illness (Fig. 243-6). Although AMI may commence at any time of the day
or night, circadian variations have been reported such that clusters are seen
in the morning within a few hours of awakening. The increased frequency early
in the day may be due to a combination of an increase in sympathetic tone and
an increased tendency to thrombosis between 6:00 A.M. and 12 noon.
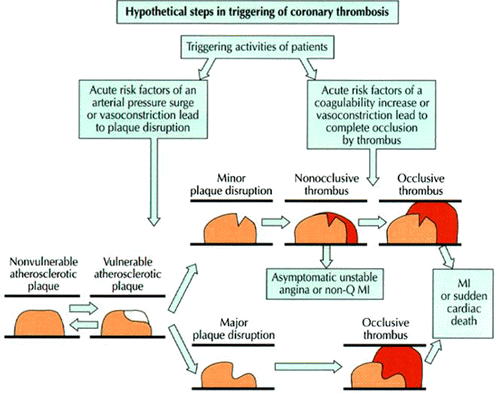
Figure 243-6: Hypothetical steps in the triggering of
coronary thrombosis. Plaque disruption with thrombus formation occurs at a
critical moment when a threshold combination of hemodynamic, prothrombotic, and
vasoconstrictive forces (acute risk factors) is rapidly generated by external
stressors (triggers) during a period of plaque vulnerability. Disruption of the
plaque may be severe enough to produce a thrombogenic stimulus sufficiently
intense to cause occlusive coronary thrombosis, leading to myocardial
infarction (MI) or sudden cardiac death.[From S Waxman, JE Muller: Risk
factors for an acute ischemic event, in RM Califf (volume ed): Acute Myocardial
Infarction and Other Acute Ischemic Syndromes, in E Braunwald (series ed):
Atlas of Heart Diseases, Philadelphia: Current Medicine, 1996, p. 2.11.]
Pain is the most common presenting complaint in patients with
AMI. In some instances, it may be severe enough to be described asthe worst
pain the patient has ever felt. The pain is deep and visceral; adjectives
commonly used to describe it are heavy, squeezing, and crushing,
although occasionally it is described as stabbing or burning (Chap. 13). It is
similar in character to the discomfort of angina pectoris but usually is more
severe and lasts longer. Typically the pain involves the central portion of the
chest and/or the epigastrium, and on occasion it radiates to the arms. Less
common sites of radiation include the abdomen, back, lower jaw, and neck. The
frequent location of the pain beneath the xiphoid and patients' denial that
they may be suffering a heart attack are chiefly responsible for the common
mistaken impression of indigestion. The pain of AMI may radiate as high as the
occipital area but not below the umbilicus. It is often accompanied by
weakness, sweating, nausea, vomiting, anxiety, and a sense of impending doom.
The pain may commence when the patient is at rest. When the pain begins during
a period of exertion, it does not usually subside with cessation of activity,
in contrast to angina pectoris.
Although
pain is the most common presenting complaint, it is by no means always present.
The proportion of painless AMIs is greater in patients with diabetes mellitus,
and it increases with age. In the elderly, AMI may present as sudden-onset
breathlessness, which may progress to pulmonary edema. Other less common
presentations, with or without pain, include sudden loss of consciousness, a
confusional state, a sensation of profound weakness, the appearance of an
arrhythmia, evidence of peripheral embolism, or merely an unexplained drop in
arterial pressure. The pain of AMI can simulate pain from acute pericarditis
(Chap. 239), pulmonary embolism (Chap. 261), acute aortic dissection (Chap.
247), costochondritis, and gastrointestinal disorders. These conditions should
therefore be considered in the differential diagnosis.
Physical Findings
Most
patients are anxious and restless, attempting unsuccessfully to relieve the
pain by moving about in bed, altering their position, and stretching. Pallor
associated with perspiration and coolness of the extremities occurs commonly.
The combination of substernal chest pain persisting for >30 min and diaphoresis
strongly suggests AMI. Although many patients have a normal pulse rate and
blood pressure within the first hour of AMI, about one-fourth of patients with
anterior infarction have manifestations of sympathetic nervous system
hyperactivity (tachycardia and/or hypertension), and up to one-half with
inferior infarction show evidence of parasympathetic hyperactivity (bradycardia
and/or hypotension).
The
precordium is usually quiet, and the apical impulse may be difficult to
palpate. In patients with anterior wall infarction, an abnormal systolic
pulsation caused by dyskinetic bulging of infarcted myocardium may develop in
the periapical area within the first days of the illness and then may resolve.
Other physical signs of ventricular dysfunction that may be present include, in
order of decreasing incidence, fourth (S4) and third (S3)
heart sounds, decreased intensity of heart sounds, and, in more severe cases,
paradoxical splitting of the second heart sound (Chap. 225). A transient apical
systolic murmur due to dysfunction of the mitral valve apparatus may be
midsystolic or late systolic in timing. A pericardial friction rub is heard in
many patients with transmural AMI at some time in the course of the disease, if
they are examined frequently. The carotid pulse is often decreased in volume,
reflecting reduced stroke volume. Jugular venous distention with clear lung
fields should raise suspicion of right ventricular infarction. Temperature
elevations up to 38°C may be observed during the first week after AMI; however,
a temperature exceeding 38°C should prompt a search for other causes. The
arterial pressure is variable; in most patients with transmural infarction,
systolic pressure declines by approximately 10 to 15 mmHg from the
preinfarction state
Laboratory Findings
Myocardial
infarction (MI) progresses through the following temporal stages: (1) acute
(first few hours to 7 days), (2) healing (7 to 28 days),and (3) healed (29 days
and beyond). When evaluating the results of diagnostic tests for AMI, the temporal
phase of the infarction process must be considered. The laboratory tests of
value in confirming the diagnosis may be divided into 4 groups: (1)
electrocardiogram (ECG), (2) serum cardiac markers, (3) cardiac imaging, and
(4) nonspecific indexes of tissue necrosis and inflammation
Electrocardiogram
The
electrocardiographic manifestations of AMI are described in Chap. 226. During
the initial stage of the acute phase of MI, total occlusion of the infarct
artery produces ST-segment elevation. Most patients initially presenting with
ST-segment elevation evolve Q waves on the ECG and are ultimately diagnosed as
having sustained a Q-wave MI. A small proportion may sustain only a non-Q-wave
MI. When the obstructing thrombus is not totally occlusive, obstruction is
transient, or if a rich collateral network is present, no ST-segment elevation
is seen. Such patients are initially considered to be experiencing either
unstable anigna or a non-ST-segment elevation MI (NSTEMI). Among patients
presenting without ST-segment elevation, if a serum cardiac marker is detected
and no Q wave develops, the diagnosis of non-Q-wave MI is ultimately made. A
minority of patients who present initially without ST-segment elevation may
develop a Q-wave MI. Previously it was believed that transmural MI is present
if the ECG demonstrates Q waves or loss of R waves, and nontransmural MI may be
present if the ECG shows only transient ST-segment and T-wave changes. However,
electrocardiographic-pathologic correlations are far from perfect; and
therefore a more rational nomenclature for designating electrocardiographic
infarction is now commonly in use, with the terms Q-wave and non-Q-wave MI
replacing the terms transmural and nontransmural MI, respectively.
The presentations that comprise the spectrum ranging from unstable angina through non-Q-wave MI to Q-wave MI are called the acute coronary syndromes (Fig. 243-1). This classification scheme provides a conceptual framework for interpreting the diagnostic and prognostic information gleaned from serum cardiac marker measurements as well as for planning antithrombotic therapy.
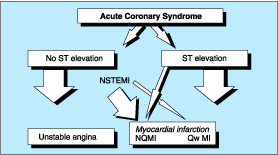
Figure 243-1: Acute coronary syndromes. Patients with
ischemic discomfort may present with or without ST-segment elevation on the
ECG. Of patients with ST-segment elevation, most (large arrow)
ultimately develop a Q-wave AMI (QwMI), while a few (small arrow)
develop a non-Q-wave AMI (NQMI). Patients who do not present with ST-segment
elevation are suffering from either unstable angina or a non-ST segment
elevation MI (NSTEMI) (large arrows), a distinction that is ultimately
made on the presence or absence of a serum cardiac marker such as CKMB or a
cardiac troponin detected in the blood. Most patients presenting with NSTEMI
ultimately develop a NQMI on the ECG; a few may develop a QwMI. The spectrum of
clinical presentations ranging from unstable angina through NQMI and QwMI are
referred to as the actue coronary syndromes.
Serum Cardiac Markers
Certain
proteins, called serum cardiac markers, are released into the blood in
large quantities from necrotic heart muscle after AMI. The rate of liberation
of specific proteins differs depending on their intracellular location and
molecular weight, and the local blood and lymphatic flow. The temporal pattern
of protein release is of diagnostic importance, but contemporary urgent
reperfusion strategies necessitate making a decision (based largely on a combination
of clinical and ECG findings) before the results of blood tests have returned
from the central laboratory. Rapid whole-blood bedside assays for serum cardiac
markers are now available and may facilitate management decisions, particularly
in patients with nondiagnostic ECGs.
Creatine
phosphokinase (CK) rises within 4 to 8 h and generally returns to normal by 48
to 72 h. An important drawback of total CK measurement is its lack of
specificity for AMI, as CK may be elevated with skeletal muscle trauma. A two-
to threefold elevation of total CK may follow an intramuscular injection, for
example. This ambiguity may lead to the erroneous diagnosis of AMI in a patient
who has been given an intramuscular injection of a narcotic for chest pain of
noncardiac origin. Other potential sources of total CK elevation are (1)
skeletal muscular diseases, including muscular dystrophy, myopathies, and
polymyositis; (2) electrical cardioversion; (3) hypothyroidism; (4) stroke; (5)
surgery; and (6) skeletal muscle damage secondary to trauma, convulsions, and
prolonged immobilization.
The
MB isoenzyme of CK has the advantage over total CK that it is not present in
significant concentrations in extracardiac tissue and therefore is considerably
more specific. However, cardiac surgery, myocarditis, and electrical
cardioversion often result in elevated serum levels of the MB isoenzyme. A
ratio (relative index) of CKMB mass:CK activity ![]() 2.5
suggests but is not diagnostic of a myocardial rather than a skeletal muscle
source for the CKMB elevation. This ratio is less useful when levels of total
CK are high owing to skeletal muscle injury or when the total CK level is
within the normal range but CKMB is elevated.
2.5
suggests but is not diagnostic of a myocardial rather than a skeletal muscle
source for the CKMB elevation. This ratio is less useful when levels of total
CK are high owing to skeletal muscle injury or when the total CK level is
within the normal range but CKMB is elevated.
Rather
than attempting to make the diagnosis of AMI on the basis of a single
measurement of CK and CKMB, clinicians should evaluate a series of measurements
obtained over the first 24 h. Skeletal muscle release of CKMB typically
produces a "plateau" pattern, whereas AMI produces a CKMB elevation
that peaks approximately 20 h after the onset of coronary occlusion. When
released into the circulation, the myocardial form of CKMB (CKMB2) is acted on
by the enzyme carboxypeptidase, which cleaves a lysine residue from the
carboxyl terminus to produce an isoform (CKMB1) with a different
electrophoretic mobility. A CKMB2:CKMB1 ratio of >1.5 is highly sensitive
for the diagnosis of AMI, particularly 4 to 6 h after the onset of coronary
occlusion.
Cardiac-specific troponin T (cTnT) and cardiac-specific
troponin I (cTnI) have amino acid sequences different from those of the
skeletal muscle forms of these proteins. These differences have permitted the
development of quantitative assays for cTnT and cTnI with highly specific
monoclonal antibodies. Since cTnT and cTnI are not normally detectable in the
blood of healthy individuals but may increase after AMI to levels over 20 times
higher than the cutoff value (usually set only slightly above the noise level
of the assay), the measurement of cTnT or cTnI is of considerable diagnostic
usefulness, and they are now the preferred biochemical markers for MI. The
cardiac troponins are particularly valuable when there is clinical suspicion of
either skeletal muscle injury or a small MI that may be below the detection
limit for CK and CKMB measurements. Levels of cTnI may remain elevated for 7 to
10 days after AMI, and cTnT levels may remain elevated for up to 10 to 14 days.
Thus, measurement of cTnT or cTnI has replaced measurement of lactate
dehydrogenase (LDH) and its isoenzymes in patients with suspected MI who come
to medical attention more than 24 to 48 h after the onset of symptoms.
Myoglobin is released into the blood within only a few
hours of the onset of AMI. Although myoglobin is one of the first serum cardiac
markers that rises above the normal range after AMI, it lacks cardiac
specificity, and it is rapidly excreted in the urine, so that blood levels
return to the normal range within 24 h of the onset of infarction.
Many
hospitals are using cTnT or cTnI rather than CKMB as the routine serum cardiac
marker for diagnosis of AMI, although any of these analytes remains clinically
acceptable. It is not cost-effective to measure both a cardiac-specific
troponin and CKMB at all time points in every patient. However, in view of the
prolonged elevation of cardiac-specific troponins (>1 week), episodes of
recurrent ischemic discomfort and suspected recurrent MI are more readily
diagnosed with a serum cardiac marker that remains elevated in the blood more
briefly, such as CKMB or myoglobin.
While
it has long been recognized that the total quantity of protein released
correlates with the size of the infarct, the peak protein concentration
correlates only weakly with infarct size. Recanalization of a coronary artery
occlusion (either spontaneously or by mechanical or pharmacologic means) in the
early hours of AMI causes earlier and higher peaking (at about 8 to 12 h after
reperfusion) of serum cardiac markers.
Characteristic
rises occur in serum cardiac markers in virtually all patients with clinically
proven MI. CK and CKMB levels generally do not rise in unstable angina.
However, approximately one-third of patients who are considered to have
unstable angina on the basis of a lack of CK or CKMB elevation have elevations
of cTnT or cTnI, probably indicating the presence of microinfarction. The
finding of an elevated cardiac-specific troponin level, even in the presence of
normal CK and CKMB values, is indicative of an adverse prognosis, and such
patients should be considered to have sustained MI and managed as described
below.
For
the purposes of confirming the diagnosis of MI, serum cardiac markers should be
measured on admission, 6 to 9 h after admission, and 12 to 24 h after admission
if the diagnosis remains uncertain.
The
nonspecific reaction to myocardial injury is associated with
polymorphonuclear leukocytosis, which appears within a few hours after the
onset of pain, persists for 3 to 7 days, and often reaches levels of 12,000 to
15,000 leukocytes per microliter. The erythrocyte sedimentation rate rises more
slowly than the white blood cell count, peaking during the first week and
sometimes remaining elevated for 1 or 2 weeks.
Cardiac Imaging
Two-dimensional echocardiography (Chap. 227) is the most
frequently employed imaging modality in patients with AMI. Abnormalities of
wall motion are almost universally present (Fig. 243-7). Even when no
ST-segment elevation is seen, echocardiographically detectable wall motion
abnormalities may be observed. Although AMI cannot be distinguished from an old
myocardial scar or from acute severe ischemia by echocardiography, the ease and
safety of the procedure make its use appealing as a screening tool. In the
emergency department setting, early detection of the presence or absence of
wall motion abnormalities by echocardiography can aid in management decisions,
such as whether the patient should receive reperfusion therapy [e.g., thrombolysis
or a percutaneous coronary intervention (PCI)]. Echocardiographic estimation of
left ventricular (LV) function is useful prognostically; detection of reduced
function serves as an indication for therapy with an angiotensin-converting
enzyme inhibitor (see "Angiotensin-Converting Enzyme Inhibitors,"
below). Echocardiography may also identify the presence of right ventricular
(RV) infarction, ventricular aneurysm, pericardial effusion, and LV thrombus.
In addition, Doppler echocardiography is useful in the detection and
quantitation of a ventricular septal defect and mitral regurgitation, two
serious complications of AMI (see below).
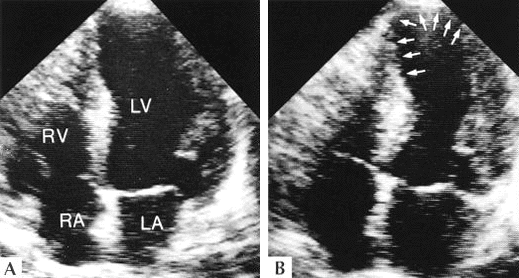
Figure 243-7: Echocardiographic wall motion abnormality
coronary artery disease. A, Two-dimensional echocardiographic apical
four-chamber view at end-diastole. B, Two-dimensional echocardiographic apical
four-chamber view at end-systole. The right ventricle (RV) and the septal and
lateral walls at the base of the left ventricle (LV) demonstrate normal inward
motion from diastole through systole; however, the distal septum and apex
demonstrate akinesis (arrows in B). The wall motion. abnormality demonstrated
in this frame was caused by ischemia from a lesion in the mid-left anterior
descending artery. LAnleft atrium; RA--right atrium. [From MH Picard, AE
Weyman: Echocardiography in chronic ischemic heart disease, in GA Beller (ed):
Chronic Ischemic Heart Disease, vol 5, in E Braunwald (series ed): Atlas of
Heart Diseases, Philadelphia: Current Medicine, 1996, pp. 4.2.]
Several
radionuclide imaging techniques are available for evaluating patients with
suspected AMI. However, these imaging modalities are used less often than
echocardiography because they are more cumbersome and they lack sensitivity and
specificity in many clinical circumstances. Myocardial perfusion imaging with 201Tl
or 99mTc-sestamibi, which are distributed in proportion to
myocardial blood flow and concentrated by viable myocardium (Chap. 244) reveal
a defect ("cold spot") in most patients during the first few hours
after development of a transmural infarct. However, although perfusion scanning
is extremely sensitive, it cannot distinguish acute infarcts from chronic scars
and thus is not specific for the diagnosis of acute MI. Radionuclide
ventriculography, carried out with 99mTc-labeled red blood cells,
frequently demonstrates wall motion disorders and reduction in the ventricular
ejection fraction in patients with AMI. While of value in assessing the
hemodynamic consequences of infarction and in aiding in the diagnosis of RV
infarction when the RV ejection fraction is depressed, this technique is also
quite nonspecific, as many cardiac abnormalities other than MI alter the
radionuclide ventriculogram.
Management
Prehospital Care
The
prognosis in AMI is largely related to the occurrence of two general classes of
complications: (1) electrical complications (arrhythmias) and (2) mechanical
problems ("pump failure"). Most out-of-hospital deaths from AMI are
due to the sudden development of ventricular fibrillation. The vast majority of
deaths due to ventricular fibrillation occur within the first 24 h of the onset
of symptoms, and, of these, over half occur in the first hour. Therefore, the
major elements of prehospital care of patients with suspected AMI include (1)
recognition of symptoms by the patient and prompt seeking of medical attention;
(2) rapid deployment of an emergency medical team capable of performing
resuscitative maneuvers, including defibrillation; and (3) expeditious
transportation of the patient to a hospital facility that is continuously
staffed by physicians and nurses skilled in managing arrhythmias, providing
advanced cardiac life support, and (4) expeditious implementation of
reperfusion therapy. The biggest delay usually occurs not during transportation
to the hospital but rather between the onset of pain and the patient's decision
to call for help. This delay can best be reduced by education of the public by
health care professionals concerning the significance of chest pain and the
importance of seeking early medical attention. Increasingly, monitoring and
treatment are carried out by trained personnel in the ambulance, further
shortening the time between the onset of the infarction and appropriate
treatment.
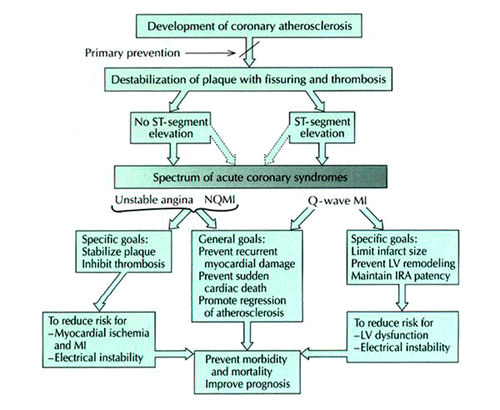
Figure 243-8: Major goals of medical therapy for acute
coronary syndromes. Once coronary atherosclerosis has developed, patients may
present with chronic exertional angina pectoris or one of the features of the
acute coronary syndromes: unstable angina pectorisjnon-Q-wave myocardial
infarction (NQMI) or Q-wave MI. A common pathophysiologic mechanism
precipitating the acute coronary syndromes is destabilization of an
atherosclerotic plaque due to fissuring of its surface.
Initial Management in the Emergency Department
In
the emergency department, the goals for the management of patients with
suspected AMI include control of cardiac pain, rapid identification of patients
who are candidates for urgent reperfusion therapy, triage of lower-risk
patients to the appropriate location in the hospital, and avoidance of
inappropriate discharge of patients with AMI. Many aspects of the treatment of
AMI are initiated in the emergency department and then continued during the
in-hospital phase of management.
Aspirin is now considered an essential element in the management of
patients with suspected AMI and is effective across the entire spectrum of
acute coronary syndromes (Fig. 243-2 and 243-3). Rapid inhibition of
cyclooxygenase in platelets followed by a reduction of thromboxane A2
levels is achieved by buccal absorption of a chewed 160 to 325 mg tablet in the
emergency department. This measure should be followed by daily oral
administration of aspirin in a dose of 160 to 325 mg.
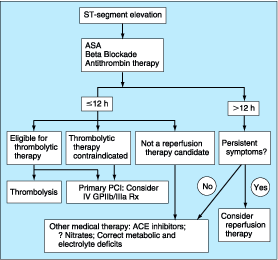
Figure 243-2: Management strategy for patients
suspected of having an ST-segment elevation. AMI patients should receive
aspirin (ASA), beta blockers (in the absence of contraindications), and an
antithrombin (particularly if a relatively fibrin-specific thrombolytic agent
is used). Adjunctive antithrombin therapy is probably not required for patients
receiving streptokinase. Patients treated within 12 h who are eligible for
thrombolytic therapy should expeditiously receive such treatment or be
considered for primary percutaneous transluminal coronary angioplasty (PCI).
Immediate, primary PCI is also to be considered when lytic therapy is
contraindicated. An intravenous glycoprotein IIb/IIIa (GPIIb/IIIa) inhibitor
may be helpful for reducing thrombotic complications during primary PCI.
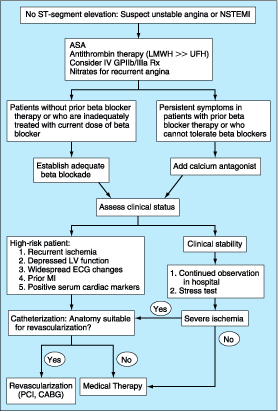
Figure 243-3: Management strategy for patients with
unstable angina and AMI without ST-segment elevation. These patients should be
treated with an antithrombin and aspirin. Nitrates should be administered for
recurrent episodes of angina. The risk of death and cardiac ischemic events may
be reduced in high risk patients if an intravenous GPIIb/IIIa inhibitor is
administered. Adequate beta blockade should be established; when that is not
possible or contraindications exist, a calcium antagonist can be considered.
Patients at high risk should be triaged to cardiac catheterization with plans
for revascularization if clinically suitable, while patients who are clinically
stable can be treated more conservatively with continued observation in the
hospital and consideration of a stress test to screen for any provocable myocardial
ischemia. CABG, coronary artery bypass grafting; LV, left ventricular.
Since
patients with AMI may develop hypoxemia secondary to ventilation-perfusion
abnormalities from LV failure and intrinsic pulmonary disease, it has been a
common practice to routinely administer supplemental oxygen. In patients
whose arterial oxygen saturation is normal as estimated by pulse oximetry or
measured by an arterial blood gas specimen, supplemental oxygen is of limited
if any clinical benefit and therefore is not cost effective. However, when
hypoxemia is present, oxygen should be administered by nasal prongs or face
mask (2 to 4 L/min) for the first 6 to 12 h after infarction; the patient
should then be reassessed to determine if there is a continued need for such
treatment.
Control of Pain
Morphine is a very effective analgesic for the pain associated with
AMI. However, it may reduce sympathetically mediated arteriolar and venous
constriction, and the resulting venous pooling may reduce cardiac output and
arterial pressure. This complication does not contraindicate the use of
morphine. Hypotension associated with venous pooling usually responds promptly
to elevation of the legs, but in some patients volume expansion with
intravenous saline is required. The patient may experience diaphoresis and
nausea, but these events usually pass and are replaced by a feeling of
well-being associated with the relief of pain. Morphine also has a vagotonic
effect and may cause bradycardia or advanced degrees of heart block,
particularly in patients with posteroinferior infarction. These side effects
usually respond to atropine (0.5 mg intravenously). Morphine is routinely
administered by repetitive (every 5 min) intravenous injection of small doses
(2 to 4 mg) rather than by the subcutaneous administration of a larger
quantity, because absorption may be unpredictable by the latter route.
Before
morphine is administered, sublingual nitroglycerin can be given safely
to most patients with AMI. Up to three 0.4-mg doses should be administered at
about 5-min intervals. In addition to diminishing or abolishing chest
discomfort, nitroglycerin, once considered contraindicated in the setting of
AMI, may be capable of both decreasing myocardial oxygen demand (by lowering
preload) and increasing myocardial oxygen supply (by dilating infarct-related
coronary vessels or collateral vessels). In patients whose initially favorable
response to sublingual nitroglycerin is followed by the return of chest pain,
particularly if accompanied by other evidence of ongoing ischemia such as
further ST-segment or T-wave shifts, the use of intravenous nitroglycerin
should be considered. Therapy with nitrates should be avoided in patients who
present with low systolic arterial pressure (<100 mmHg) or in whom there is
clinical suspicion of right ventricular infarction (inferior infarction on
electrocardiogram, elevated jugular venous pressure, clear lungs, and
hypotension). Nitrates should not be administered to patients who have taken
the phosphodiasterase 5 inhibitor sildenafil for erectile dysfunction within
the preceding 24 h since it may potentiate the hypotensive effects of nitrates.
An idiosyncratic reaction to nitrates, consisting of sudden marked hypotension,
sometimes occurs but can usually be reversed promptly by the rapid
administration of intravenous atropine.
Intravenous
beta blockers are also useful in the control of the pain of AMI. These
drugs control pain effectively in some patients, presumably by diminishing
myocardial oxygen demand and hence ischemia. More important, there is evidence
that intravenous beta blockers reduce in-hospital mortality, particularly in
high-risk patients (see "![]() -Adrenoceptor
Blockers," below). A commonly employed regimen is metoprolol, 5 mg every 2
to 5 min for a total of three doses, provided the patient has a heart rate
>60 beats per minute (bpm), systolic pressure >100 mmHg, a PR interval
<0.24 s, and rales that are no higher than 10 cm up from the diaphragm.
Fifteen minutes after the last intravenous dose, an oral regimen is initiated
of 50 mg every 6 h for 48 h followed by 100 mg every 12 h.
-Adrenoceptor
Blockers," below). A commonly employed regimen is metoprolol, 5 mg every 2
to 5 min for a total of three doses, provided the patient has a heart rate
>60 beats per minute (bpm), systolic pressure >100 mmHg, a PR interval
<0.24 s, and rales that are no higher than 10 cm up from the diaphragm.
Fifteen minutes after the last intravenous dose, an oral regimen is initiated
of 50 mg every 6 h for 48 h followed by 100 mg every 12 h.
Unlike
beta blockers, calcium antagonists are of little value in the acute setting,
and there is evidence that short-acting dihydropyridines may be associated with
an increased mortality risk.
Management Strategies (Figs. 243-2
and 243-3)
The
primary tool for screening patients and making triage decisions is the initial
12-lead ECG. When ST-segment elevation in at least two contiguous leads of at
least 2 mm in V1-V3 and 1 mm in other leads is present, a patient should be
considered a candidate for reperfusion therapy (Fig. 243-2 (Fig. 243-9).
If no contraindications are present (see "Contraindications and
Complications," under "Thombolysis," below), thrombolytic
therapy should ideally be initiated within 30 min. The process of selecting
patients for thrombolysis versus primary PCI (angioplasty, or stenting) (Chap.
245) is discussed below. In the absence of ST-segment elevation, thrombolysis
is not helpful, and evidence exists suggesting that it may be harmful.
Pharmacotherapy for patients presenting without ST-segment elevation (Fig.
243-3) typically includes measures to control cardiac pain (as discussed
above), aspirin, antithrombin therapy (preferably with low-molecular-weight
heparin), and infusion of nitroglycerin as needed to control recurrent
ischemia. For high-risk patients an intravenous infusion of a glycoprotein
IIb/IIIa inhibitor should be considered. Further management recommendations for
patients without ST-segment elevation are outlined in Fig. 243-3.
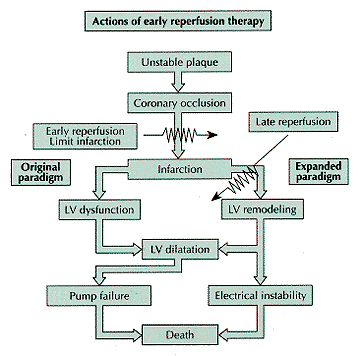
Figure 243-9: Expansion of the paradigm of the
beneficial actions of early reperfusion therapy. The original paradigm appears
on the left and the expanded paradigm is on the right. Restoration of coronary
flow during the early (salvage) phase of acute myocardial infarction (MI) is
administered to reduce MI size. Recent observations of the influence of infarct
vessel patency on left ventricular (LV) remodeling and electrical stability led
to an expansion of the original paradigm. [From MA Pfeffer: Cardiac
remodeling and its prevention, in WS Colucci: Heart Failure: Cardiac Function
and Dysfunction, in E Braunwald (series ed): Atlas of Heart Diseases, vol 4.
Philadelphia: Current Medicine, 1995, p. 5.11. Adapted from C Kim, E Braunwald:
Potential benefits of late reperfusion of infarcted myocardium: the open artery
hypothesis. Circulation 88:2426-2436, 1993.]
Limitation of Infarct Size
The
quantity of myocardium that becomes necrotic as a consequence of a coronary
artery occlusion is determined by factors other than just the site of
occlusion. While the central zone of the infarct contains necrotic tissue that
is irretrievably lost, the fate of the surrounding ischemic myocardium may be
improved by timely restoration of coronary perfusion, reduction of myocardial
oxygen demands, prevention of the accumulation of noxious metabolites, and
blunting of the impact of mediators of reperfusion injury (e.g., calcium
overload and oxygen-derived free radicals). Up to one-third of patients with
AMI may achieve spontaneous reperfusion of the infarct-related coronary
artery within 24 h and experience improved healing of infarcted tissue.
Reperfusion either pharmacologically (by thrombolysis) or mechanically (by
angioplasty and/or stenting) accelerates the process of opening the occluded
infarct-related artery in those patients in whom spontaneous thrombolysis
ultimately would have occurred and also greatly increases the number of
patients in whom restoration of flow in the infarct-related artery is
accomplished. Timely restoration of flow in the epicardial infarct-related
artery combined with improved perfusion of the downstream zone of infarcted
myocardium results in a limitation of infarct size. Protection of the ischemic
myocardium by the maintenance of an optimal balance between myocardial oxygen
supply and demand through pain control, treatment of congestive heart failure,
and minimization of tachycardia and hypertension extends the "window"
of time for the salvage of myocardium by reperfusion strategies.
Glucocorticoids
and nonsteroidal anti-inflammatory agents, with the exception of aspirin,
should be avoided in the setting of AMI. They can impair infarct healing and
increase the risk of myocardial rupture, and their use may result in a larger
infarct scar. In addition, they can increase coronary vascular resistance,
thereby potentially reducing flow to ischemic myocardium.
Thrombolysis
The
thrombolytic agents tissue plasminogen activator (tPA), streptokinase,
anisoylated plasminogen streptokinase activator complex (APSAC) and reteplase
(rPA) have been approved by the Food and Drug Administration for intravenous
use in the setting of AMI. These drugs all act by promoting the conversion of
plasminogen to plasmin, which subsequently lyses fibrin thrombi. Although
considerable emphasis was first placed on a distinction between more
fibrin-specific agents, such as tPA, and non-fibrin-specific agents, such as
streptokinase, it is now recognized that these differences are only relative,
as some degree of systemic fibrinolysis occurs with tPA. The principal goal of
thrombolysis is prompt restoration of coronary arterial patency.
When
assessed angiographically, flow in the culprit coronary artery is described by
a simple qualitative scale called the TIMI grading system: grade 0 indicates
complete occlusion of the infarct-related artery; grade 1 indicates some
penetration of the contrast material beyond the point of obstruction but
without perfusion of the distal coronary bed; grade 2 indicates perfusion of
the entire infarct vessel into the distal bed but with flow that is delayed
compared with that of a normal artery; and grade 3 indicates full perfusion of
the infarct vessel with normal flow. Early reports frequently lumped TIMI
grades 2 and 3 under the general category of patency, but it is now
recognized that grade 3 flow is the goal of reperfusion therapy, because full
perfusion of the infarct-related coronary artery yields far better results in
terms of infarct size, maintenance of LV function, and reduction of both short-
and long-term mortality rates. Relatively new methods of angiographic
assessment of the efficacy of thrombolysis include counting the number of
frames required on the cine film for dye to flow from the origin of the
infarct-related artery to a landmark in the distal vascular bed (TIMI frame
count) and determining the rate of entry and exit of contrast dye from the
microvasculature in the myocardial infarct zone (TIMI Myocardial Perfusion
Grade).
Thrombolytic
therapy can reduce the relative risk of in-hospital death by up to 50% when
administered within the first hour of the onset of symptoms of AMI, and much of
this benefit is maintained for at least 10 years. Appropriately used
thrombolytic therapy appears to reduce infarct size, limit LV dysfunction, and
reduce the incidence of serious complications such as septal rupture,
cardiogenic shock, and malignant ventricular arrhythmias. Since myocardium can
be salvaged only before it has been irreversibly injured, the timing of
reperfusion therapy, by thrombolysis or a catheter-based approach, is of
extreme importance in achieving maximum benefit. While the upper time limit
depends on specific factors in individual patients, it is clear that
"every minute counts" and that patients treated within 1 to 3 h of
the onset of symptoms generally benefit most. Although reduction of the
mortality rate is more modest, the therapy remains of benefit for many patients
seen 3 to 6 h after the onset of infarction, and some benefit appears to be
possible up to 12 h, especially if chest discomfort is still present and ST
segments remain elevated in ECG leads that do not yet demonstrate new Q waves.
In addition to the possibility of early treatment, clinical factors that favor
proceeding with thrombolytic therapy include anterior wall injury,
hemodynamically complicated infarction, and widespread ECG evidence of
myocardial jeopardy. Although patients (younger than 65 years) achieve a
greater relative reduction in the mortality rate than elderly patients, the higher
absolute mortality rate (15 to 25%) in elderly patients results in
similar absolute reductions in the mortality rates for both age groups.
Intriguing
data are accumulating to indicate that improved ventricular function and
reduced mortality may also be achieved by late coronary reperfusion. The
benefits of late reperfusion cannot be attributed to a reduction of infarct
size but appear to result from improvement of tissue healing in the infarct
zone with prevention of infarct expansion, enhancement of collateral flow,
improvement of myocardial contractile performance, and reduction in the
tendency to electrical instability. In addition, hibernating myocardium
(i.e., poorly contractile myocardium in a zone that is supplied by a stenotic
infarct-related coronary artery with slow antegrade perfusion, Chap. 244) may
show improved contraction after angioplasty to increase coronary blood flow.
tPA
is more effective than streptokinase at restoring full perfusion-i.e., TIMI
grade 3 coronary flow-and has a small edge in improving survival as well. The
current recommended regimen of tPA consists of a 15-mg bolus followed by 50 mg
intravenously over the first 30 min, followed by 35 mg over the next 60 min.
Streptokinase is administered as 1.5 million units (MU) intravenously over 1 h.
Reteplase is administered in a double bolus regimen consisting of a 10-MU bolus
given over 2 to 3 min followed by a second 10-MU bolus 30 min later.
Promising
new pharmacologic regimens for reperfusion combine an intravenous glycoprotein
IIb/IIIa inhibitor with a reduced dose of a thrombolytic agent. Such
combination reperfusion regimens appear to facilitate the rate and extent of
thrombolysis by inhibiting platelet aggregation, weakening the clot structure,
and allowing penetration of the thrombolytic agent deeper into the clot.
Contraindications and Complications
Clear
contraindications to the use of thrombolytic agents include a history of
cerebrovascular hemorrhage at any time, a nonhemorrhagic stroke or other
cerebrovascular event within the past year, marked hypertension (a reliably
determined systolic arterial pressure >180 mmHg and/or a diastolic pressure
>110 mmHg) at any time during the acute presentation, suspicion of aortic
dissection, and active internal bleeding (excluding menses). While advanced age
is associated with an increase in hemorrhagic complications, the benefit of
thrombolytic therapy in the elderly appears to justify its use if no other
contraindications are present and the amount of myocardium in jeopardy appears to
be substantial.
Relative
contraindications to thrombolytic therapy, which require careful assessment of
the risk:benefit ratio, include current use of anticoagulants (international
normalized ratio ![]() 2),
a recent (<2 weeks) invasive or surgical procedure or prolonged (>10 min)
cardiopulmonary resuscitation, known bleeding diasthesis, pregnancy, a
hemorrhagic ophthalmic condition (e.g., hemorrhagic diabetic retinopathy),
active peptic ulcer disease, and a history of severe hypertension that is
currently adequately controlled. Because of the risk of an allergic reaction,
patients should not receive streptokinase if that agent had been received
within the preceding 5 days to 2 years.
2),
a recent (<2 weeks) invasive or surgical procedure or prolonged (>10 min)
cardiopulmonary resuscitation, known bleeding diasthesis, pregnancy, a
hemorrhagic ophthalmic condition (e.g., hemorrhagic diabetic retinopathy),
active peptic ulcer disease, and a history of severe hypertension that is
currently adequately controlled. Because of the risk of an allergic reaction,
patients should not receive streptokinase if that agent had been received
within the preceding 5 days to 2 years.
Allergic reactions to streptokinase occur in approximately 2% of
patients who receive it. While a minor degree of hypotension occurs in 4 to 10%
of patients given this agent, marked hypotension occurs, although rarely, in
association with severe allergic reactions.
Hemorrhage is the most frequent and potentially the most
serious complication. Because bleeding episodes that require transfusion are
more common when patients require invasive procedures, unnecessary venous or
arterial interventions should be avoided in patients receiving thrombolytic
agents. Hemorrhagic stroke is the most serious complication and occurs in
approximately 0.5 to 0.9% of patients being treated with these agents. This rate
increases with advancing age, with patients older than 70 years experiencing
roughly twice the rate of intracranial hemorrhage as those younger than 65
years. Large-scale intervention trials have suggested that the rate of
intracranial hemorrhage with tPA or rPA is slightly higher than that with
streptokinase.
Routine angiography after thrombolysis with the intent of
performing a PCI on underlying coronary artery stenoses in the culprit vessel
is not recommended. Higher rates of abrupt closure of the infarct-related
coronary artery with a need for urgent coronary artery bypass surgery as well
as a trend toward an increase in mortality rate have been noted with this
approach. Instead, after thrombolytic therapy, cardiac catheterization and
coronary angiography should be carried out if there is evidence of either (1)
failure of reperfusion (persistent chest pain and ST-segment elevation beyond
90 min) in which case a rescue PCI should be considered, or (2) coronary
artery reocclusion (reelevation of ST segments and/or recurrent chest pain) or
the development of recurrent ischemia (such as recurrent angina in the early
hospital course or a positive exercise stress test before discharge), in which
case an elective PCI should be considered. Coronary artery bypass
surgery should be reserved for patients whose coronary anatomy is unsuited to
angioplasty but in whom revascularization appears to be advisable because of
extensive jeopardized myocardium or recurrent ischemia.
Primary Percutaneous Coronary Intervention
PCI,
usually angioplasty and/or stenting without preceding thrombolysis, is also
effective in restoring perfusion in AMI when carried out on an emergency basis
in the first few hours of MI. It has the advantage of being applicable to
patients who have contraindications to thrombolytic therapy but otherwise are
considered appropriate candidates for reperfusion. It appears to be more
effective than thrombolysis in opening occluded coronary arteries and, when
performed by experienced operators in dedicated medical centers, is
associated with better short-term and long-term clinical outcomes. It remains
to be determined whether the advantages of primary PCI reported from organized
research efforts can be replicated in routine clinical practice. However, PCI is
expensive in terms of personnel and facilities, and its applicability is
seriously limited by its availability, around the clock, in only a minority of
hospitals.
Hospital Phase Management
Coronary Care Units
These
units are routinely equipped with a system that permits continuous monitoring
of the cardiac rhythm of each patient and hemodynamic monitoring in selected
patients. Defibrillators, respirators, noninvasive transthoracic pacemakers,
and facilities for introducing pacing catheters and flow-directed
balloon-tipped catheters are also usually available. Equally important is the
organization of a highly trained team of nurses who can recognize arrhythmias;
adjust the dosage of antiarrhythmic, vasoactive, and anticoagulant drugs; and
perform cardiac resuscitation, including electroshock, when necessary.
Patients
should be admitted to a coronary care unit early in their illness when it is
expected that they will derive benefit from the sophisticated and expensive
care provided. The availability of electrocardiographic monitoring and trained
personnel outside the coronary care unit has made it possible to admit
lower-risk patients (e.g., those not hemodynamically compromised and without
active arrhythmias) to "intermediate care units."
The
duration of stay in the coronary care unit is dictated by the ongoing need for
intensive care. If AMI has been ruled out (ideally within 8 to 12 h) and
symptoms are controlled with oral therapy, patients may be transferred out of
the coronary care unit. Also, patients who have a confirmed AMI but who are
considered to be at low risk (no prior infarction and no persistent chest
discomfort, congestive heart failure, hypotension, or cardiac arrhythmias) may
be safely transferred out of the coronary care unit in 24 to 36 h.
Activity
Factors
that increase the work of the heart during the initial hours of infarction may
increase the size of the infarct. Therefore, patients with AMI should be kept
at bed rest for the first 12 h. However, in the absence of complications,
patients should be encouraged, under supervision, to resume an upright posture
by dangling their feet over the side of the bed and sitting in a chair within
the first 24 h. This practice is both psychologically beneficial and usually
results in a reduction in the pulmonary capillary wedge pressure. In the
absence of hypotension and other complications, by the second or third day
patients typically are ambulating in their room with increasing duration and
frequency, and they may shower or stand at the sink to bathe. By day 3 or 4
after infarction, patients should be increasing their ambulation progressively
to a goal of 600 ft at least three times a day.
Diet
Because
of the risk of emesis and aspiration soon after MI, patients should receive
either nothing or only clear liquids by mouth for the first 4 to 12 h. The
typical coronary care unit diet should provide ![]() 30%
of total calories as fat and have a cholesterol content of
30%
of total calories as fat and have a cholesterol content of ![]() 300
mg/d. Complex carbohydrates should make up 50 to 55% of total calories.
Portions should not be unusually large, and the menu should be enriched with
foods that are high in potassium, magnesium, and fiber but low in sodium.
Diabetes mellitus and hypertriglyceridemia are managed by restriction of
concentrated sweets in the diet.
300
mg/d. Complex carbohydrates should make up 50 to 55% of total calories.
Portions should not be unusually large, and the menu should be enriched with
foods that are high in potassium, magnesium, and fiber but low in sodium.
Diabetes mellitus and hypertriglyceridemia are managed by restriction of
concentrated sweets in the diet.
Bowels
Bed
rest and the effect of the narcotics used for the relief of pain often lead to
constipation. A bedside commode rather than a bedpan, a diet rich in bulk, and
the routine use of a stool softener such as dioctyl sodium sulfosuccinate (200
mg/d) are recommended. If the patient remains constipated despite these
measures, a laxative can be prescribed. Contrary to prior belief, it is safe to
perform a gentle rectal examination on patients with AMI.
Sedation
Many
patients require sedation during hospitalization to withstand the period of
enforced inactivity with tranquillity. Diazepam (5 mg), oxazepam (15 to 30 mg),
or lorazepam (0.5 to 2 mg), given three or four times daily, is usually
effective. An additional dose of any of the above medications may be given at
night to ensure adequate sleep. Attention to this problem is especially
important during the first few days in the coronary care unit, where the
atmosphere of 24-h vigilance may interfere with the patient's sleep. However,
sedation is no substitute for reassuring, quiet surroundings. Many drugs used
in the coronary care unit, such as atropine, H2 blockers, and
narcotics, can produce delirium, particularly in the elderly. This effect
should not be confused with agitation, and it is wise to conduct a thorough
review of the patient's medications before arbitrarily prescribing additional
doses of anxiolytics.
Pharmacotherapy
Antithrombotic Agents
The
use of antiplatelet and antithrombin therapy during the initial phase of AMI is
based on extensive laboratory and clinical evidence that thrombosis plays an
important role in the pathogenesis of this condition. The primary goal of
treatment with antiplatelet and antithrombin agents is to establish and
maintain patency of the infarct-related artery. A secondary goal is to reduce
the patient's tendency to thrombosis and thus the likelihood of mural thrombus
formation or deep venous thrombosis, either of which could result in pulmonary
embolization. The degree to which antiplatelet and antithrombin therapy
achieves these goals partly determines how effectively it reduces the risk of
mortality from AMI.
As
noted previously (see "Initial Management in the Emergency
Department," above), aspirin is the standard antiplatelet agent for
patients with AMI. The most compelling evidence for the benefits of
antiplatelet therapy (mainly with aspirin) in AMI is found in the comprehensive
overview by the Antiplatelet Trialists' Collaboration. Data from nearly 20,000
patients with AMI enrolled in nine randomized trials were pooled and revealed a
reduction in the mortality rate from 11.7% in control patients to 9.3% in
patients receiving antiplatelet agents. This difference corresponds to the
prevention of 24 deaths for every 1000 patients treated. Similarly, 2 strokes and
12 recurrent infarctions are prevented for every 1000 patients treated with
antiplatelet therapy.
The
glycoprotein IIb/IIIa receptor is the focus of intense investigation by basic
and clinical scientists (Fig. 243-10)(Chap. 116). Because platelet-rich thrombi
are more resistant to thrombolytic agents than platelet-poor thrombi and
because platelet aggregates appear to play a role in reocclusion after
initially successful thrombolysis, glycoprotein IIb/IIIa inhibition may
facilitate thrombolysis and reduce the rate of reocclusion of reperfused
vessels. Compounds have been developed that block the glycoprotein IIb/IIIa
receptor. These drugs appear useful for preventing thrombotic complications in
patients with AMI undergoing PCI and reduce the rate of the composite endpoint
of death and recurrent AMI in the medical management of patients without
ST-segment elevation at presentation.
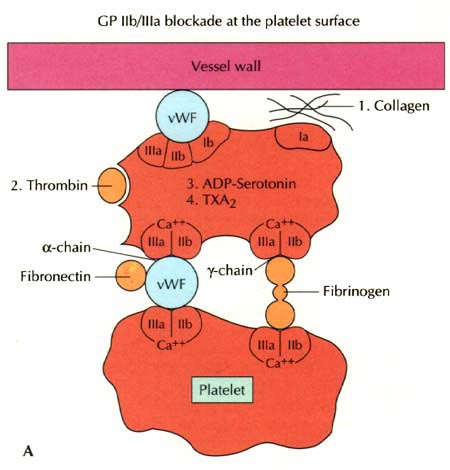
Figure 243-10: Despite its
effectiveness, aspirin is a relatively weak platelet antagonist and does not
effectively inhibit the platelet aggregation caused by many of the stimuli
thought to be important in active ischemic heart disease (e. g., thrombin).
The
standard antithrombin agent used in clinical practice is unfractionated heparin
(UFH). Despite numerous clinical trials, the precise role of heparin in
patients treated with thrombolytic agents remains uncertain. The available data
fail to show any convincing benefit of UFH with respect to either coronary
arterial patency or mortality rate when UFH is added to a regimen of aspirin
and a non-fibrin-specific thrombolytic agent such as streptokinase. Although
not conclusively proven, it appears that the immediate administration of
intravenous UFH, in addition to a regimen of aspirin and tPA, helps to
facilitate thrombolysis and to establish and maintain patency of the
infarct-related artery. This effect is achieved at the cost of a small
increased risk of bleeding. Most clinicians who use tPA also administer a bolus
and infusion of UFH, which should be administered as a bolus of 60 U/kg
followed by a maintenance infusion of 12 U/kg per hour. The activated partial
thromboplastin time during maintenance therapy should be 1.5 to 2 times the
control value.
An
alternative to UFH for anticoagulation of patients with AMI that is being used
with increased frequency are the low-molecular-weight heparin preparations
(LMWHs), which are formed by enzymatic or chemical depolymerization to produce
saccharide chains of varying length but with a mean molecular weight of about
5000 Da. The LMWHs have several advantages over UFH including an increased
anti-factor Xa:IIa ratio, decreased sensitivity to platelet factor IV, a more stable
reliable anticoagulant effect, and enhanced bioavailability, thereby permitting
administration via the subcutaneous route. Because of the stable anticoagulant
effect when LMWHs are used, routine monitoring of hematologic tests such as the
activated partial thromboplastin time (aPTT) is not required. Although the
LMWHs share many pharmacologic similarities, they also vary in a number of
important features; and therefore these agents should be considered
individually rather than as members of an interchangeable class of compounds.
Of the LMWHs, nadroparin and dalteparin have been found to be similar to UFH in
therapeutic effectiveness, while enoxaparin (1 mg/kg subcutaneously every 12 h)
appears to be superior to UFH for reducing the mortality rate and cardiac
ischemic events in patients with AMI who do not present with ST-segment
elevation. Direct comparisons among the LMWHs have not been carried out.
Patients
with an anterior location of the infarction, severe LV dysfunction, congestive
heart failure, a history of embolism, two-dimensional echocardiographic
evidence of mural thorombus, or atrial fibrillation are at increased risk of
systemic or pulmonary thromboembolism. Such individuals should receive full
therapeutic levels of antithrombin therapy (UFH or LMWHs) while hospitalized,
followed by at least 3 months of warfarin therapy.
Beta-Adrenoceptor Blockers
The
benefits of beta blockers in patients with AMI can be divided into those that
occur immediately when the drug is given acutely and those that accrue over the
long term when the drug is given for secondary prevention after an index
infarction. Acute intravenous beta blockade improves the myocardial oxygen
supply-demand relationship, decreases pain, reduces infarct size, and decreases
the incidence of serious ventricular arrhythmias. An overview of the data from
27,000 patients enrolled in nine randomized trials in the prethrombolytic era
indicates that intravenous followed by oral beta blockade is associated with a
15% relative reduction in mortality, nonfatal reinfarction, and nonfatal
cardiac arrest. In patients who undergo thrombolysis soon after the onset of
chest pain, no incremental reduction in mortality rate is seen with beta
blockers, but recurrent ischemia and reinfarction are reduced.
Beta
blocker therapy after AMI thus is useful for most patients except those in whom
it is specifically contraindicated (patients with heart failure or severely
compromised LV function, heart block, orthostatic hypotension, or a history of
asthma) and perhaps those whose excellent long-term prognosis (defined as an
expected mortality rate of <1% per year) markedly diminishes any potential
benefit (patients younger than 55 years with normal ventricular function, no
complex ventricular ectopy, and no angina).
Although
the data supporting the use of beta blockers in patients with AMI who do not
present with ST-segment elevation are limited, the available evidence suggests
that even among such patients, the use of beta blockers decreases the rates of
cardiovascular mortality and reinfarction, and increases the probability of
long-term survival.
Angiotensin Converting Enzyme Inhibitors
Angiotensin-converting
enzyme (ACE) inhibitors reduce the mortality rate after AMI, and the mortality
benefits are additive to those achieved with aspirin and beta blockers. The
maximum benefit is seen in high-risk patients (those who are elderly or have an
anterior infarction, a prior infarction, and/or globally depressed LV
function), but evidence suggests that a short-term benefit occurs when ACE
inhibitors are prescribed unselectively to all hemodynamically stable patients
with AMI (i.e., those with a systolic pressure >100 mmHg). The mechanism
involves a reduction in ventricular remodeling after infarction (see
"Ventricular Dysfunction," below) with a subsequent reduction in the
risk of congestive heart failure (CHF). The rate of recurrent infarction also
may be lower in patients treated chronically with ACE inhibitors after
infarction (Fig. 243-11).
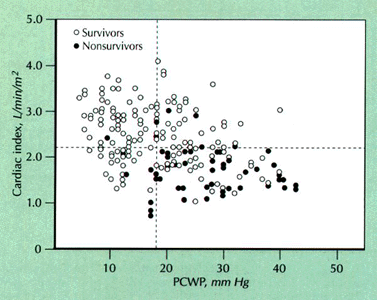
Figure 243-11: Cardiac index versus
pulmonary capillary wedge pressure (PCWP) in 200 patients after acute
myocardial infarction (MI), demonstrating an inverse relationship between
mortality and cardiac performance.
ACE
inhibitors should be prescribed within 24 h to all patients with AMI and overt
CHF as well as to hemodynamically stable patients with ST-segment elevation or
left bundle branch block. There is little evidence to support the immediate use
of ACE inhibitors in patients with AMI who present without ST-segment changes
or only with ST-segment depression without CHF. Before hospital discharge, LV
function should be assessed with an imaging study. ACE inhibitors should be
continued indefinitely in patients who have clinically evident CHF, in patients
whom an imaging study shows a reduction in global LV function or a large
regional wall motion abnormality, or in those who are hypertensive.
Other Agents
Although
the actual impact on the mortality rate is slight (three to four lives saved
per 1000 patients treated), nitrates (intravenous or oral) may be useful
in the relief of pain associated with AMI. Favorable effects on the ischemic
process and ventricular remodeling (see below) have led many physicians to
routinely use intravenous nitroglycerin (5 to 10 ![]() g/min
initial dose and up to 200
g/min
initial dose and up to 200 ![]() g/min
as long as hemodynamic stability is maintained) for the first 24 to 48 h after
the onset of infarction.
g/min
as long as hemodynamic stability is maintained) for the first 24 to 48 h after
the onset of infarction.
Results
of multiple trials of different calcium antagonists have failed to establish a
role for these agents in the treatment of most patients with AMI, in contrast
to the more consistent data that exist for other drugs (e.g., beta blockers,
aspirin, thrombolytic agents). The routine use of calcium antagonists cannot be
recommended.
A
metabolic supportive measure that has shown promise in several small-scale
trials of patients with AMI is the administration of a solution of
glucose-insulin-potassium (GIK). A GIK infusion lowers the concentration of
plasma free fatty acids and improves ventricular performance. Strict control of
blood glucose in diabetic patients with AMI has been shown to reduce the
mortality rate. It remains to be determined whether infusions of GIK should be
administered to all patients with AMI.
Intracellular
magnesium levels are frequently reduced in patients with AMI, but this
deficit is not adequately reflected in serum measurements, as magnesium is
predominantly an intracellular ion and <1% of its total body stores is
intravascular. Whether giving routine empirical supplemental infusions of
magnesium to high-risk patients with AMI is beneficial remains an open
question. At present, serum magnesium should be measured in all patients on
admission, and any demonstrated deficits should be corrected to minimize the
risk of arrhythmias. There does not appear to be any benefit in the routine use
of magnesium when it is administered relatively late (after more than 6 h) or
to patients with an uncomplicated AMI who have a low mortality risk. Its role
in high-risk patients is under investigation.
Complications and Their Treatment
Ventricular Dysfunction
After
AMI, the LV undergoes a series of changes in shape, size, and thickness in both
the infarcted and noninfarcted segments. This process is referred to as ventricular
remodeling and generally precedes the development of clinically evident CHF
in the months to years after infarction. Soon after AMI, the LV begins to
dilate. Acutely, this results from expansion of the infarct (i.e., slippage of
muscle bundles, disruption of normal myocardial cells, and tissue loss within
the necrotic zone, resulting in disproportionate thinning and elongation of the
infarct zone). Later, lengthening of the noninfarcted segments occurs as well.
The overall chamber enlargement that occurs is related to the size and location
of the infarct, with greater dilation following infarction of the apex of the
LV and causing more marked hemodynamic impairment, more frequent heart failure,
and a poorer prognosis. Progressive dilation and its clinical consequences may
be ameliorated by therapy with ACE inhibitors and other vasodilators (e.g.,
nitrates) (Fig. 243-12). Thus, in patients with an ejection fraction <40%,
regardless of whether or not heart failure is present, ACE inhibitors should be
prescribed.
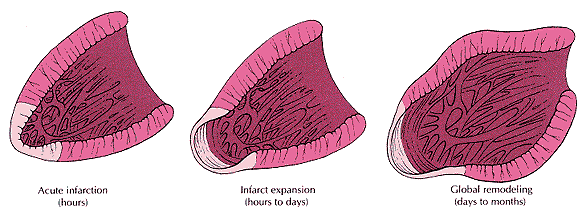
Figure 243-12: Left ventricular
remodeling after myocardial infarction (MI).
Hemodynamic Assessment
Pump
failure is now the primary cause of in-hospital death from AMI. The extent of
ischemic necrosis correlates well with the degree of pump failure and with
mortality, both early (within 10 days of infarction) and later. The most common
clinical signs are pulmonary rales and S3 and S4 gallop
rhythms. Pulmonary congestion is also frequently seen on the chest
roentgenogram. Elevated LV filling pressure and elevated pulmonary artery
pressure are the characteristic hemodynamic findings, but these findings may
result from a reduction of ventricular compliance (diastolic failure) and/or a
reduction of stroke volume with secondary cardiac dilation (systolic failure)
(Chap. 231).
A
classification originally proposed by Killip divides patients into four groups:
class I, no signs of pulmonary or venous congestion; class II, moderate heart
failure as evidenced by rales at the lung bases, S3 gallop,
tachypnea, or signs of failure of the right side of the heart, including venous
and hepatic congestion; class III, severe heart failure, pulmonary edema; and
class IV, shock with systolic pressure <90 mmHg and evidence of peripheral
vasoconstriction, peripheral cyanosis, mental confusion, and oliguria. When
this classification was established in 1967, the expected hospital mortality
rate of patients in these classes was as follows: class I, 0 to 5%; class II,
10 to 20%; class III, 35 to 45%; and class IV, 85 to 95%. With advances in
management, the mortality rate in each class has fallen, perhaps by as much as
one-third to one-half.
Hemodynamic
evidence of abnormal LV function appears when contraction is seriously impaired
in 20 to 25% of the LV. Infarction of ![]() 40%
of the LV usually results in cardiogenic shock (see below). Positioning of a
balloon flotation catheter in the pulmonary artery permits monitoring of LV
filling pressure; this technique is useful in patients who exhibit hypotension
and/or clinical evidence of CHF (Fig. 243-13). Cardiac output can also be
determined with a pulmonary artery catheter. With the addition of intraarterial
pressure monitoring, systemic vascular resistance can be calculated as a guide
to adjusting vasopressor and vasodilator therapy. Some patients with AMI have
markedly elevated LV filling pressures (>22 mmHg) and normal cardiac indexes
[>2.6 and >3.6 L/(min/m2)], while others have relatively low
LV filling pressures (<15 mmHg) and reduced cardiac indexes. The former
patients usually benefit from diuresis, while the latter may respond to volume
expansion by means of intravenous administration of colloid-containing
solutions.
40%
of the LV usually results in cardiogenic shock (see below). Positioning of a
balloon flotation catheter in the pulmonary artery permits monitoring of LV
filling pressure; this technique is useful in patients who exhibit hypotension
and/or clinical evidence of CHF (Fig. 243-13). Cardiac output can also be
determined with a pulmonary artery catheter. With the addition of intraarterial
pressure monitoring, systemic vascular resistance can be calculated as a guide
to adjusting vasopressor and vasodilator therapy. Some patients with AMI have
markedly elevated LV filling pressures (>22 mmHg) and normal cardiac indexes
[>2.6 and >3.6 L/(min/m2)], while others have relatively low
LV filling pressures (<15 mmHg) and reduced cardiac indexes. The former
patients usually benefit from diuresis, while the latter may respond to volume
expansion by means of intravenous administration of colloid-containing
solutions.
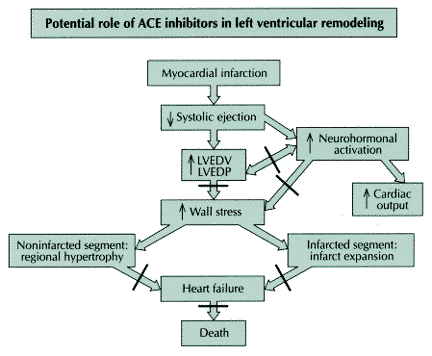
Figure 243-13: Shown with intersecting
bars are sites at which an ACE inhibitor may interrupt an adverse consequence
of myocardial infarction that would contribute to heart failure or death.
Hypovolemia
Hypovolemia
is an easily corrected condition that may contribute to the hypotension and
vascular collapse associated with AMI in some patients. It may be secondary to
previous diuretic use, to reduced fluid intake during the early stages of the
illness, and/or to vomiting associated with pain or medications. Consequently,
hypovolemia should be identified and corrected in patients with AMI and
hypotension before more vigorous forms of therapy are begun. Central venous pressure
reflects RV rather than LV filling pressure and is an inadequate guide for
adjustment of blood volume, since LV function is almost always affected much
more adversely than RV function in patients with AMI. The optimal LV filling or
pulmonary artery wedge pressure may vary considerably among patients. Each
patient's ideal level (generally ~20 mmHg) is reached by cautious fluid
administration during careful monitoring of oxygenation and cardiac output.
Eventually, the cardiac output level plateaus, and further increases in LV
filling pressure only increase congestive symptoms and decrease systemic
oxygenation without raising arterial pressure.
![]() Treatment
Treatment
The
management of CHF in association with AMI is similar to that of acute heart
failure secondary to other forms of heart disease (avoidance of hypoxemia,
diuresis, afterload reduction, inotropic support) (Chap. 232), except that the
benefits of digitalis administration to patients with AMI are unimpressive. By
contrast, diuretic agents are extremely effective, as they diminish pulmonary
congestion in the presence of systolic and/or diastolic heart failure. Left
ventricular filling pressure falls and orthopnea and dyspnea improve after the
intravenous administration of furosemide or other loop diuretics. These drugs
should be used with caution, however, as they can result in a massive diuresis
with associated decreases in plasma volume, cardiac output, systemic blood
pressure, and hence coronary perfusion. Nitrates in various forms may be used
to decrease preload and congestive symptoms. Oral isosorbide dinitrate, topical
nitroglycerin ointment, or intravenous nitroglycerin all have the advantage
over a diuretic of lowering preload through venodilation without decreasing the
total plasma volume. In addition, nitrates may improve ventricular compliance
if ischemia is present, as ischemia causes an elevation of LV filling pressure.
The patient with pulmonary edema is treated as described in Chap. 232, but
vasodilators must be used with caution to prevent serious hypotension. As noted
earlier, ACE inhibitors are an ideal class of drugs for management of
ventricular dysfunction after AMI, especially for the long term.
Cardiogenic Shock
In
recent years, efforts to reduce infarct size and prompt treatment of ongoing
ischemia and other complications of MI appear to have reduced the incidence of
cardiogenic shock from 20% to about 7%. Only 10% of patients with this
condition present with it on admission, while 90% develop it during
hospitalization. Typically, patients who develop cardiogenic shock have severe
multivessel coronary artery disease with evidence of "piecemeal"
necrosis extending outward from the original infarct zone (Fig. 243-4).
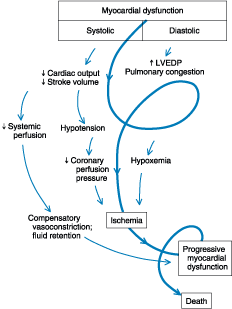
Figure 243-4: Pathophysiology of cardiogenic shock:
Systolic and diastolic myocardial dysfunction occurs after MI, resulting in
pulmonary congestion and a reduction in cardiac output. Systemic and coronary
hypoperfusion occur, resulting in progressive ischemia. Although a number of
compensatory mechanisms are activated in an attempt to support the circulation,
these compensatory mechanisms may become maladaptive and produce a worsening of
hemodynamics. A vicious circle of progressive myocardial dysfunction occurs
that ultimately results in death if it is not interrupted. LVEDP, left
ventricular end-diastolic pressure.
Cardiogenic
shock should be considered to be a form of severe LV failure. This syndrome is
characterized by marked hypotension with systolic arterial pressure of <80
mmHg and a markedly reduced cardiac index [<1.8 L/(min/m2)] in
the face of an elevated LV filling (pulmonary capillary wedge) pressure (>18
mmHg). Hypotension alone is not a basis for the diagnosis of cardiogenic shock,
because many patients who make an uneventful recovery have serious hypotension
(systolic pressure of <80 mmHg) for several hours. Such patients often have
low LV filling pressures, and their hypotension usually resolves with the
administration of intravenous fluids. In contrast to hypovolemic hypotension,
cardiogenic shock is generally associated with a mortality rate of >70%;
however, recent efforts to restore perfusion by coronary angioplasty or
surgical revascularization suggest that this high mortality rate can be lowered
by as much as one-half.
Risk
factors for the in-hospital development of shock include advanced age, a
depressed LV ejection fraction on admission, a large infarct, previous MI, and
a history of diabetes mellitus. Patients with several of these risk factors
should be considered for cardiac catheterization and mechanical reperfusion (by
PCI or surgery) before the development of shock.
Pathophysiology of Severe Power Failure
A
marked reduction in the quantity of contracting myocardium is the cause of
cardiogenic shock in AMI. The initial insult reduces arterial pressure, and the
reduction in coronary perfusion pressure and myocardial blood flow initiates a
vicious cycle that impairs myocardial function further and may increase the
size of the infarct (Fig. 243-4). Arrhythmias and metabolic acidosis also
contribute to this deterioration, because they are the result of inadequate
perfusion. This positive feedback loop accounts for the high mortality rate
associated with the shock syndrome.
Right Ventricular Infarction
Approximately
one-third of patients with inferoposterior infarction demonstrate at least a
minor degree of RV necrosis. An occasional patient with inferoposterior LV
infarction also has extensive RV infarction, and rare patients present with
infarction limited primarily to the RV. Clinically significant RV infarction
causes signs of severe RV failure [jugular venous distention, Kussmaul's sign
(Chap. 225), hepatomegaly] with or without hypotension. ST-segment elevations
of right-sided precordial ECG leads, particularly lead V4R, are
frequently present in the first 24 h in patients with RV infarction.
Two-dimensional echocardiography is helpful in determining the degree of RV
dysfunction. Catheterization of the right side of the heart often reveals a
distinctive hemodynamic pattern resembling cardiac tamponade or constrictive
pericarditis (steep right atrial "y" descent and an early diastolic
dip and plateau in right ventricular waveforms) (Chap. 239). Therapy consists
of volume expansion to maintain adequate RV preload and efforts to improve LV
performance with attendant reduction in pulmonary capillary wedge and pulmonary
arterial pressures.
Mechanical Causes of Heart Failure
Free Wall Rupture
Myocardial
rupture is a dramatic complication of AMI that is most likely to occur during
the first week after the onset of symptoms; its frequency increases with the
age of the patient. First infarction, a history of hypertension, no history of
angina pectoris, and a relatively large Q-wave infarct are associated with a
higher incidence of cardiac rupture. The clinical presentation typically is a sudden
loss of pulse, blood pressure and consciousness while the ECG continues to show
sinus rhythm (apparent electromechanical dissociation or pulseless electrical
activity). The myocardium continues to contract, but forward flow is not
maintained as blood escapes into the pericardium. Cardiac tamponade (Chap. 239)
ensues, and closed-chest massage is ineffective. This condition is almost
universally fatal, although dramatic cases of urgent pericardiotensis followed
by successful surgical repair have been reported.
Ventricular Septal Defect
The
pathogenesis of perforation of the ventricular septum is similar to that of
free wall rupture, but the chance of successful therapy is greater. Patients
with ventricular septal rupture present with sudden, severe LV failure in
association with the appearance of a pansystolic murmur, often accompanied by a
parasternal thrill. It is often impossible to differentiate this condition from
rupture of a papillary muscle with resulting mitral regurgitation (MR), and the
presence in both conditions of a tall "v" wave in the pulmonary
capillary wedge pressure further complicates the differentiation. The diagnosis
of ventricular septal defect can be established by the demonstration of a
left-to-right shunt (i.e., an oxygen step-up at the level of the RV) by means
of limited cardiac catheterization performed at the bedside with a
flow-directed balloon catheter. Color flow Doppler echocardiography can also be
extremely useful for making this diagnosis at the bedside (Fig. 243-14). A prolonged
period of hemodynamic compromise may produce end-organ damage and other
complications that can be avoided by early intervention, including
nitroprusside infusion and intraaortic balloon counterpulsation.
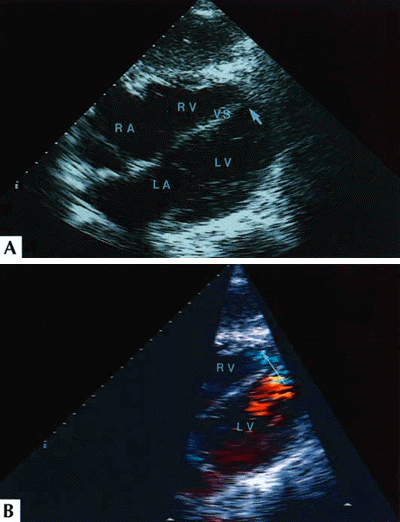
Figure 243-14: Two-dimensional
echocardiogram demonstrating ventricular septal (VS) rupture (VSR) following
acute myocardial infarction.
The
pathophysiology of acute MR is similar to that of acute ventricular septal
perforation in that the level of aortic systolic pressure partly determines the
regurgitant volume, the principal difference being the chamber into which the
regurgitant fraction is ejected. In septal perforation, a fraction of LV output
is ejected into the right ventricle. As in MR, lowering of the aortic systolic
pressure by mechanical (intraaortic balloon counterpulsation) and/or
pharmacologic (nitroglycerin or nitroprusside) means can decrease the
hemodynamic compromise caused by perforation.
Mitral Regurgitation
The
reported incidence of apical systolic murmurs of MR during the first few days
after the onset of AMI varies widely (from 10 to 50% of patients) depending on
the population studied and the acumen of the observers. While MR causes acute
hemodynamic compromise in only a minority of these patients, it is a risk
factor for late CHF and reduced survival.
The
most common cause of MR after AMI is dysfunction of the mitral valve due to
ischemia or infarction. Left ventricular dilatation or alteration in the size
or shape of the LV due to impaired contractility or to aneurysm formation
causes disordered contraction of the papillary muscles and failure of
coaptation of the mitral valve leaflets. Rarely, a papillary muscle, or, more
commonly, the head of a papillary muscle, may rupture. Then, LV function
deteriorates dramatically, with superimposition of severe MR. The major element
in the differential diagnosis is perforation of the ventricular septum as discussed
above. Surgical repair or replacement of the mitral valve may lead to dramatic
improvement in patients in whom acute heart failure results primarily from
severe MR due to papillary muscle rupture or dysfunction and in whom global
ventricular function is relatively good.
If
aortic systolic pressure is lowered in patients with MR, a greater fraction of
the LV output will be ejected antegrade, thus lessening the regurgitant
fraction. To this end, both intraaortic balloon counterpulsation (IABC), which
lowers the aortic systolic pressure mechanically, and the infusion of
nitroglycerin or sodium nitroprusside, which reduce systemic vascular
resistance, have been used with success in the interim management of patients
with severe MR in the presence of AMI. Ideally, definitive operative treatment
should be postponed until pulmonary congestion has cleared and the infarct has
had time to heal. However, if the patient's hemodynamic and/or clinical
condition does not improve or stabilize, surgical treatment should be
undertaken, even in the acute stage.
Arrhythmias
The
incidence of arrhythmias after AMI is higher in patients seen early after the
onset of symptoms. The mechanisms responsible for infarction-related
arrhythmias include autonomic nervous system imbalance, electrolyte
disturbances, ischemia, and slowed conduction in zones of ischemic myocardium.
An arrhythmia can usually be managed successfully if trained personnel and
appropriate equipment are available when it develops. Since most deaths from
arrhythmia occur during the first few hours after infarction, the effectiveness
of treatment relates directly to the speed with which patients come under
medical observation. The prompt management of arrhythmias constitutes a
significant advance in the treatment of myocardial infarction.
Ventricular Premature Beats
Infrequent,
sporadic ventricular premature depolarizations occur in almost all patients
with AMI and do not require therapy. Whereas in the past, frequent, multifocal,
or early diastolic ventricular extrasystoles (so-called warning arrhythmias)
were routinely treated with antiarrhythmic drugs to reduce the risk of
development of ventricular tachycardia and ventricular fibrillation,
pharmacologic therapy is now reserved for patients with sustained ventricular
arrhythmias. Prophylactic antiarrhythmic therapy (either intravenous lidocaine
early or oral agents later) is contraindicated for ventricular premature beats
in the absence of clinically important ventricular tachyarrhythmias, as such
therapy may actually increase the mortality rate. ![]() -Adrenoceptor
blocking agents are effective in abolishing ventricular ectopic activity in
patients with AMI and in the prevention of ventricular fibrillation. As
described above (see "
-Adrenoceptor
blocking agents are effective in abolishing ventricular ectopic activity in
patients with AMI and in the prevention of ventricular fibrillation. As
described above (see "![]() -Adrenoceptor
Blockers"), they should be used routinely in patients without
contraindications. In addition, hypokalemia and hypomagnesemia are risk factors
for ventricular fibrillation in patients with AMI; the serum potassium
concentration should be adjusted to approximately 4.5 mmol/L and magnesium to
about 2.0 mmol/L.
-Adrenoceptor
Blockers"), they should be used routinely in patients without
contraindications. In addition, hypokalemia and hypomagnesemia are risk factors
for ventricular fibrillation in patients with AMI; the serum potassium
concentration should be adjusted to approximately 4.5 mmol/L and magnesium to
about 2.0 mmol/L.
Ventricular Tachycardia and Fibrillation
Within
the first 24 h of AMI, ventricular tachycardia and fibrillation can occur
without prior warning arrhythmias. The occurrence of ventricular fibrillation
can be reduced by prophylactic administration of intravenous lidocaine.
However, prophylactic use of lidocaine has not been shown to reduce overall
mortality from AMI. In fact, in addition to causing possible noncardiac
complications, lidocaine may predispose to an excess risk of bradycardia and
asystole. For these reasons, and with earlier treatment of active ischemia,
more frequent use of beta-blocking agents, and the nearly universal success of
electrical cardioversion or defibrillation, routine prophylactic antiarrhythmic
drug therapy is no longer recommended. It should be reserved for patients who
cannot reach a hospital or for those treated in hospitals that lack the
constant presence in the coronary care unit of a physician or nurse trained in
the recognition and treatment of ventricular fibrillation.
Sustained
ventricular tachycardia that is well tolerated hemodynamically should be
treated with an intravenous regimen of lidocaine (bolus of 1.0 to 1.5 mg/kg;
infusion of 20 to 50 ![]() g/kg
per min), procainamide (bolus of 15 mg/kg over 20 to 30 min; infusion of 1 to 4
mg/min), or amiodarone (bolus of 75 to 150 mg over 10 to 15 min followed by
infusion of 1.0 mg/min for 6 h and then 0.5 mg/min); if it does not stop
promptly, electroversion should be used (Chap. 230). An unsynchronized
discharge of 200 to 300 J (defibrillation) is used immediately in patients with
ventricular fibrillation or when ventricular tachycardia causes hemodynamic
deterioration. Ventricular tachycardia or fibrillation that is refractory to
electroshock may be more responsive after the patient is treated with
epinephrine (1 mg intravenously or 10 mL of a 1:10,000 solution via the
intracardiac route), bretylium (a 5 mg/kg bolus), or amiodarone (a 75 to 150 mg
bolus).
g/kg
per min), procainamide (bolus of 15 mg/kg over 20 to 30 min; infusion of 1 to 4
mg/min), or amiodarone (bolus of 75 to 150 mg over 10 to 15 min followed by
infusion of 1.0 mg/min for 6 h and then 0.5 mg/min); if it does not stop
promptly, electroversion should be used (Chap. 230). An unsynchronized
discharge of 200 to 300 J (defibrillation) is used immediately in patients with
ventricular fibrillation or when ventricular tachycardia causes hemodynamic
deterioration. Ventricular tachycardia or fibrillation that is refractory to
electroshock may be more responsive after the patient is treated with
epinephrine (1 mg intravenously or 10 mL of a 1:10,000 solution via the
intracardiac route), bretylium (a 5 mg/kg bolus), or amiodarone (a 75 to 150 mg
bolus).
Ventricular
arrhythmias, including the unusual form of ventricular tachycardia known as torsade
de pointes (Chap. 230), may occur in patients with AMI as a consequence of
other concurrent problems (such as hypoxia, hypokalemia, or other electrolyte
disturbances) or of the toxic effects of an agent being administered to the
patient (such as digoxin or quinidine). A search for such secondary causes should
always be undertaken.
Although
the in-hospital mortality rate is increased, the long-term survival is good in
patients who survive to hospital discharge after primary ventricular
fibrillation, i.e., ventricular fibrillation that is a primary response to
acute ischemia and is not associated with predisposing factors such as CHF,
shock, bundle branch block, or ventricular aneurysm. This result is in sharp
contrast to the poor prognosis for patients who develop ventricular
fibrillation secondary to severe pump failure. For patients who develop
ventricular tachycardia or ventricular fibrillation late in their hospital
course (i.e., after the first 48 h), the mortality rate is increased both
in-hospital and during long-term follow-up. Such patients should be considered
for electrophysiologic study (Chap. 230).
Accelerated Idioventricular Rhythm
Accelerated
idioventricular rhythm (AIVR, "slow ventricular tachycardia"), a
ventricular rhythm with a rate of 60 to 100 beats per minute, occurs in 25% of
patients with AMI. It often occurs transiently during thrombolytic therapy at
the time of reperfusion. The rate of AIVR is usually similar to that of the
sinus rhythm that precedes and follows it, and this similarity of rate plus the
relatively minor hemodynamic effects make this rhythm more difficult to detect
except by electrocardiographic monitoring. For the most part, AIVR is benign
and does not presage the development of classic ventricular tachycardia. Most
episodes of AIVR do not require treatment if the patient is monitored
carefully, as degeneration into a more serious arrhythmia is rare, and, if it
occurs, AIVR can generally be readily treated with a drug that increases the
sinus rate (atropine).
Supraventricular Arrhythmias
Sinus
tachycardia is the most common supraventricular arrhythmia. If it occurs
secondary to another cause (such as anemia, fever, heart failure, or a
metabolic derangement), the primary problem should be treated first. However,
if it appears to be due to sympathetic overstimulation, for example, as part of
a hyperdynamic state, then treatment with a beta blocker is indicated. Other
common arrhythmias in this group are atrial flutter and atrial fibrillation,
which are often secondary to LV failure. Digoxin is usually the treatment of
choice for supraventricular arrhythmias if heart failure is present. If heart
failure is absent, beta blockers, verapamil, or diltiazem are suitable
alternatives for controlling the ventricular rate, as they may also help to
control ischemia. If the abnormal rhythm persists for >2 h with a
ventricular rate in excess of 120 beats per minute, or if tachycardia induces
heart failure, shock, or ischemia (as manifested by recurrent pain or ECG
changes), a synchronized electroshock (100 to 200 J) should be used.
Accelerated
junctional rhythms have diverse causes but may occur in patients with
inferoposterior infarction. Digitalis excess must be ruled out. In some
patients with severely compromised LV function, the loss of appropriately timed
atrial systole results in a marked decrease in cardiac output. Right atrial or
coronary sinus pacing is indicated in such instances.
Sinus Bradycardia
Treatment
of sinus bradycardia is indicated if hemodynamic compromise results from the
slow heart rate. Atropine is the most useful drug for increasing heart rate and
should be given intravenously in doses of 0.5 mg initially. If the rate remains
below 50 to 60 bpm, additional doses of 0.2 mg, up to a total of 2.0 mg, may be
given. Persistent bradycardia (<40 bpm) despite atropine may be treated with
electrical pacing. Isoproterenol should be avoided.
Atrioventricular and Intraventricular Conduction
Disturbances
Both
the in-hospital mortality rate and the post-discharge mortality rate of
patients who have complete atrioventricular (AV) block in association with
anterior infarction are markedly higher than those of patients who develop AV
block with inferior infarction. This difference is related to the fact that
heart block in inferior infarction is commonly a result of increased vagal tone
and/or the release of adenosine and therefore is transient. In anterior wall
infarction, heart block is usually related to ischemic malfunction of the
conduction system, which commonly is associated with extensive myocardial
necrosis.
Temporary
electrical pacing provides an effective means of increasing the heart rate of
patients with bradycardia due to AV block. However, acceleration of the heart
rate may have only a limited impact on prognosis in patients with anterior wall
infarction and complete heart block in whom the large size of the infarct is
the major factor determining outcome. It should be carried out if it improves
hemodynamics, however. Pacing does appear to be beneficial in patients with
inferoposterior infarction who have complete heart block associated with heart
failure, hypotension, marked bradycardia, or significant ventricular ectopic
activity. A subgroup of these patients, those with RV infarction, often respond
poorly to ventricular pacing because of the loss of the atrial contribution to
ventricular filling. In such patients, dual-chamber AV sequential pacing may be
required.
External
noninvasive pacing electrodes should be positioned in a "demand" mode
for patients with sinus bradycardia (rate <50 bpm) that is unresponsive to
drug therapy, Mobitz II second-degree AV block, third-degree heart block, or
bilateral bundle branch block (e.g., right bundle branch block plus left
anterior fascicular block). Retrospective studies suggest that permanent pacing
may reduce the long-term risk of sudden death due to bradyarrhythmias in the
rare patient who develops combined persistent bifascicular and transient
third-degree heart block during the acute phase of MI.
Other Complications
Recurrent Chest Discomfort
Recurrent
angina develops in ~25% of patients hospitalized for AMI. This percentage is
even higher in patients who undergo successful thrombolysis. Since recurrent or
persistent ischemia often heralds extension of the original infarct or
reinfarction in a new myocardial zone and is associated with a doubling of risk
after AMI, patients with these symptoms should be considered for repeat
thrombolysis or referred for prompt coronary arteriography and mechanical
revascularization. Repeat administration of a thrombolytic agent is an
alternative to early mechanical revascularization.
Pericarditis
Pericardial
friction rubs and/or pericardial pain are frequently encountered in patients
with transmural AMI. This complication can usually be managed with aspirin (650
mg qid). It is important to diagnose the chest pain of pericarditis accurately,
since failure to recognize it may lead to the erroneous diagnosis of recurrent
ischemic pain and/or infarct extension, with resulting inappropriate use of
anticoagulants, nitrates, beta blockers, or coronary arteriography. When it
occurs, complaints of pain radiating to either trapezius muscle is helpful
since such a pattern of discomfort is typical of pericarditis but rarely occurs
with ischemic discomfort. Anticoagulants potentially could cause tamponade in
the presence of acute pericarditis (as manifested by either pain or persistent
rub) and therefore should not be used unless there is a compelling indication.
Thromboembolism
Clinically
apparent thromboembolism complicates AMI in ~10% of cases, but embolic lesions
are found in 20% of patients in necropsy series, suggesting that
thromboembolism is often clinically silent. Thromboembolism is considered to be
at least an important contributing cause of death in 25% of patients with AMI
who die after admission to the hospital. Arterial emboli originate from LV
mural thrombi, while most pulmonary emboli arise in the leg veins.
Thromboembolism
typically occurs in association with large infarcts (especially anterior), CHF,
and a LV thrombus detected by echocardiography. The incidence of arterial
embolism from a clot originating in the ventricle at the site of an infarction
is small but real. Two-dimensional echocardiography reveals LV thrombi in about
one-third of patients with anterior wall infarction but in few patients with
inferior or posterior infarction. Arterial embolism often presents as a major
complication, such as hemiparesis when the cerebral circulation is involved or
hypertension if the renal circulation is compromised. When a thrombus has been
clearly demonstrated by echocardiographic or other techniques or when a large
area of regional wall motion abnormality is seen even in the absence of a
detectable mural thrombus, systemic anticoagulation should be undertaken (in
the absence of contraindications), as the incidence of embolic complications
appears to be markedly lowered by such therapy. The appropriate duration of
therapy is unknown, but 3 to 6 months is probably prudent.
Left Ventricular Aneurysm
The
term ventricular aneurysm is usually used to describe dyskinesis
or local expansile paradoxical wall motion. Normally functioning myocardial
fibers must shorten more if stroke volume and cardiac output are to be
maintained in patients with ventricular aneurysm; if they cannot, overall
ventricular function is impaired. True aneurysms are composed of scar tissue
and neither predispose to nor are associated with cardiac rupture.
The
complications of LV aneurysm do not usually occur for weeks to months after
AMI; they include CHF, arterial embolism, and ventricular arrhythmias. Apical
aneurysms are the most common and the most easily detected by clinical
examination. The physical finding of greatest value is a double, diffuse, or
displaced apical impulse. Ventricular aneurysms are readily detected by
two-dimensional echocardiography, which may also reveal a mural thrombus in an
aneurysm.
Rarely,
myocardial rupture may be contained by a local area of pericardium, along with
organizing thrombus and hematoma. Over time, this pseudoaneurysm
enlarges, maintaining communication with the LV cavity through a narrow neck.
Because a pseudoaneurysm often ruptures spontaneously, it should be surgically
repaired if recognized.
Postinfarction Risk Stratification and
Management
Many
clinical factors have been identified that are associated with an increase in
cardiovascular risk after initial recovery from AMI. Some of the most important
factors include persistent ischemia (spontaneous or provoked), depressed LV
ejection fraction (<40%), rales above the lung bases on physical examination
or congestion on chest radiograph, and symptomatic ventricular arrhythmias.
Other features associated with increased risk include a history of previous
myocardial infarction, age over 70 years, diabetes, prolonged sinus
tachycardia, hypotension, ST-segment changes at rest without angina
("silent ischemia"), an abnormal signal-averaged ECG, nonpatency of
the infarct-related coronary artery (if angiography is undertaken), and
persistent advanced heart block or a new intraventricular conduction abnormality
on the ECG. Therapy must be individualized on the basis of the relative
importance of the risk(s) present.
The
goal of preventing reinfarction and death after recovery from AMI has led to
strategies to evaluate risk after infarction. Early after AMI, this evaluation
generally involves the use of noninvasive testing. In stable patients,
submaximal exercise stress testing may be carried out before hospital discharge
to detect residual ischemia and ventricular ectopy and to provide the patient
with a guideline for exercise in the early recovery period. Alternatively, or
in addition, a maximal (symptom-limited) exercise stress test may be carried
out 4 to 6 weeks after infarction. Evaluation of LV function at rest and during
exercise is usually warranted as well. Recognition of a depressed LV ejection
fraction by echocardiography or radionuclide ventriculography identifies
patients who should receive ACE inhibitors (see "Angiotensin-Converting
Enzyme Inhibitors," above). Patients in whom angina is induced at relatively
low workloads, those who have a large reversible defect on perfusion imaging or
a depressed ejection fraction, those with demonstrable ischemia, and those in
whom exercise provokes symptomatic ventricular arrhythmias should be considered
at high risk for recurrent MI or death from arrhythmia; and cardiac
catheterization with coronary angiography and/or invasive electrophysiologic
evaluation is advised.
Exercise
tests also aid in formulating an individualized exercise prescription, which
can be much more vigorous in patients who tolerate exercise without any of the
above-mentioned adverse signs. Additionally, predischarge stress testing may
provide an important psychological benefit, building the patient's confidence
by demonstrating a reasonable exercise tolerance. Furthermore, particularly
when no arrhythmias or signs of ischemia are identified, the patient benefits
by the physician's reassurance that objective evidence suggests no immediate
jeopardy.
In
many hospitals a cardiac rehabilitation program with progressive exercise is
initiated in the hospital and continued after discharge. Ideally, such programs
should include an educational component that informs patients about their
disease and its risk factors.
The
usual duration of hospitalization for an uncomplicated AMI is about 5 days. The
remainder of the convalescent phase may be accomplished at home. During the
first 2 weeks, the patient should be encouraged to increase activity by walking
about the house and outdoors in good weather. Normal sexual activity may be
resumed during this period. After 2 weeks, the physician must regulate the
patient's activity on the basis of exercise tolerance. Most patients will be
able to return to work within 2 to 4 weeks.
Secondary Prevention of Infarction
Various
secondary preventive measures are at least partly responsible for the
improvement in the long-term mortality and morbidity rates after AMI. Long-term
treatment with an antiplatelet agent (usually aspirin) after AMI is associated
with a 25% reduction in the risk of recurrent infarction, stroke, or
cardiovascular mortality (36 fewer events for every 1000 patients treated). In
addition, in patients taking aspirin chronically, AMIs tend to be smaller and
are more likely to be non-Q-wave in nature. An alternative antiplatelet agent
that may be used for secondary prevention in patients intolerant of aspirin is
the ADP receptor antagonist clopidogrel (75 mg orally daily). ACE inhibitors
should be used indefinitely by patients with clinically evident heart failure,
a moderate decrease in global ejection fraction, or a large regional wall
motion abnormality to prevent late ventricular remodeling and recurrent
ischemic events.
The
chronic routine use of oral ![]() -adrenoceptor
blockers for at least 2 years after AMI is supported by well-conducted,
placebo-controlled trials that have convincingly demonstrated reductions in the
rates of total mortality, sudden death, and, in some instances, reinfarction.
In contrast, calcium antagonists are not recommended for routine secondary
prevention.
-adrenoceptor
blockers for at least 2 years after AMI is supported by well-conducted,
placebo-controlled trials that have convincingly demonstrated reductions in the
rates of total mortality, sudden death, and, in some instances, reinfarction.
In contrast, calcium antagonists are not recommended for routine secondary
prevention.
Evidence
suggests that warfarin lowers the risk of late mortality and the incidence of
reinfarction after AMI. Since studies comparing aspirin and warfarin therapy
separately or in combination have not yet been completed, most physicians
prescribe aspirin routinely for all patients without contraindications and add
warfarin for patients at increased risk of embolism (see
"Thromboembolism," above).
Finally,
risk factors for atherosclerosis (Chap. 241) should be discussed with
the patient, and, when possible, favorably modified. In particular, efforts
should be made to ensure the cessation of smoking and the control of
hypertension and hyperlipidemia (the target low-density lipoprotein level is
<100 mg/dL). In addition, regular physical exercise and reduction of
emotional stress should be encouraged. The benefits of hormone replacement
therapy in postmenopausal women recovering from MI remain controversial. The
initiation of a combination of estrogen plus progestin is associated with an
increased risk of cardiovascular events within the first year but may reduce
events in later years (HERS Trial). Thus, hormone replacement therapy
prevention of coronary events should not be given de novo to
postmenopausal women after AMI. Postmenopausal women already taking estrogen
plus progestin at the time of AMI may continue that therapy.