HEART FAILURE
Heart
failure (HF) is the pathophysiologic state in which an abnormality of cardiac
function is responsible for the failure of the heart to pump blood at a rate
commensurate with the requirements of the me tabolizing tissues and/or
allows it to do so only from an abnormally elevated diastolic volume. HF is
frequently, but not always, caused by a defect in myocardial contraction, and
then the term myocardial failure is appropriate. The latter may result
from a primary abnormality in heart muscle, as occurs in the cardiomyopathies,
in viral myocarditis (Chap. 238), and with excessive programmed cell death
(apoptosis). HF also results commonly from coronary atherosclerosis, which
interferes with cardiac contraction by causing myocardial infarction and
ischemia. HF may also occur in valvular and/or congenital heart disease in
which the heart muscle is damaged by the long-standing excessive hemodynamic
burden imposed by the valvular abnormality or cardiac malformation.
In
other patients with HF, however, a similar clinical syndrome is present but
without any detectable abnormality of myocardial function. In some of
these patients the normal heart is suddenly presented with a mechanical load
that exceeds its capacity, such as an acute hypertensive crisis, rupture of an
aortic valve cusp, or massive pulmonary embolism. HF in the presence of normal
myocardial function also occurs in chronic conditions in which there is
impaired filling of the ventricles due to a mechanical abnormality such as
tricuspid and/or mitral stenosis, constrictive pericarditis without myocardial
involvement, endocardial fibrosis, and some forms of hypertrophic
cardiomyopathy. In many patients with HF, particularly those with valvular or
congenital heart disease, there is a combination of impaired myocardial
function and hemodynamic overload.
Heart
failure should be distinguished from (1) conditions in which there is
circulatory congestion secondary to abnormal salt and water retention but in
which there is no disturbance of cardiac function per se, as occurs in renal
failure; and (2) noncardiac causes of inadequate cardiac output, such as
hypovolemic shock (Chap. 38).
The
ventricles respond to a chronically increased hemodynamic burden with the
development of hypertrophy (Fig. 232-1). When the ventricle is called on to
deliver an elevated cardiac output for prolonged periods, as in valvular
regurgitation, it develops eccentric hypertrophy, i.e., cavity
dilatation, with an increase in muscle mass so that the ratio between wall
thickness and ventricular cavity size remains relatively constant early in the
process. With chronic pressure overload, as in valvular aortic stenosis or
untreated hypertension, concentric ventricular hypertrophy develops; in
this condition the ratio between wall thickness and ventricular cavity size
increases. In both eccentric and concentric hypertrophy, a stable
hyperfunctioning state may exist for many years, but myocardial function may
ultimately deteriorate, leading to HF. Often at this time, the ventricle dilates
and the ratio between wall thickness and cavity size declines, leading to
increased stress on each unit of myocardium, further dilatation, and a vicious
circle.
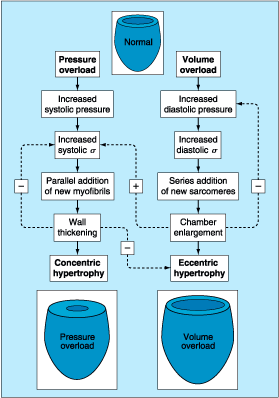
Figure 232-1: Patterns of ventricular hypertrophy.
Specific patterns of ventricular remodeling occur in response to the imposed
augmentation in work load. A pattern of hypertrophic growth characterized as
concentric, in which increased mass is out of proportion to chamber volume, is
particularly effective in reducing systolic wall stress (![]() )
under conditions of heightened pressure load. In contrast, in volume overload
conditions, in which the major stimulus is diastolic loading, a predominant
finding is a great increase in the cavity size or volume. Although there can be
extensive increases in mass, the relationship between mass and volume is either
preserved or, in severe cases, reduced. The fundamental response is generated
by cellular hypertrophy. However, the configuration of the new contractile
tissue is specific and offsets the mechanical stimulus.
)
under conditions of heightened pressure load. In contrast, in volume overload
conditions, in which the major stimulus is diastolic loading, a predominant
finding is a great increase in the cavity size or volume. Although there can be
extensive increases in mass, the relationship between mass and volume is either
preserved or, in severe cases, reduced. The fundamental response is generated
by cellular hypertrophy. However, the configuration of the new contractile
tissue is specific and offsets the mechanical stimulus.
Heart
failure represents a major public health problem in industrialized nations. It
appears to be the only common cardiovascular condition that is increasing in
prevalence and incidence. In the United States, HF is responsible for almost 1
million hospital admissions and 40,000 deaths annually. Since HF is more common
in the elderly, its prevalence is likely to continue to increase as the
population ages.
Causes
of Heart Failure
In
evaluating patients with HF, it is important to identify not only the underlying
but also the precipitating cause. The cardiac abnormality produced by a
congenital or acquired lesion such as valvular aortic stenosis may exist for
many years and cause no clinical disability. Frequently, however, clinical
manifestations of HF are precipitated for the first time in the course of some
acute disturbance that places an additional load on a myocardium that is
chronically excessively burdened (see below). Such a heart may be compensated
but have little additional reserve, and the additional load imposed by a
precipitating cause results in further deterioration of cardiac function.
Identification of such precipitating causes is of critical importance because
their prompt alleviation may be lifesaving. In the absence of underlying heart
disease, these acute disturbances do not by themselves lead to HF.
Precipitating Causes
Infection. Patients with pulmonary vascular congestion
due to left ventricular failure are more susceptible to pulmonary infection
than are normal persons; any infection may precipitate HF. The resulting fever,
tachycardia, and hypoxemia and the increased metabolic demands may place a
further burden on an overloaded, but compensated, myocardium of a patient with
chronic heart disease.
Anemia. In the presence of anemia, the oxygen needs of the
metabolizing tissues can be met only by an increase in the cardiac output Although
such an increase in cardiac output can be sustained by a normal heart, a
diseased, overloaded, but otherwise compensated heart may be unable to augment
sufficiently the volume of blood that it delivers to the periphery. In this
manner, the combination of anemia and previously compensated heart disease can
precipitate HF and lead to inadequate delivery of oxygen to the periphery.
Thyrotoxicosis and pregnancy. Similar to anemia and
fever, thyrotoxicosis and pregnancy are also high cardiac output states. The
development or intensification of HF in a patient with previously compensated
heart disease may actually be one of the first clinical manifestations of
hyperthyroidism. Similarly, HF not infrequently occurs for the first time
during pregnancy in women with rheumatic valvular disease, in whom cardiac
compensation may return following delivery .
Arrhythmias. In patients with compensated heart disease,
arrhythmias are among the most frequent precipitating causes of HF. They exert
a deleterious effect for a variety of reasons: (a) Tachyarrhythmias reduce the
time period available for ventricular filling and in patients with ischemic
heart disease they may also cause ischemic myocardial dysfunction. (b) The
dissociation between atrial and ventricular contractions characteristic of many
brady- and tachyarrhythmias results in the loss of the atrial booster pump mechanism,
thereby raising atrial pressures. (c) Cardiac performance may become further
impaired because of the loss of normally synchronized ventricular contraction
in any arrhythmia associated with abnormal intraventricular conduction. (d)
Marked bradycardia associated with complete atrioventricular block or other
severe bradyarrhythmias reduces cardiac output unless stroke volume rises
reciprocally; this compensatory response cannot occur with serious myocardial
dysfunction, even in the absence of HF).
Rheumatic, viral, and other forms of myocarditis. Acute rheumatic fever
and a variety of other inflammatory or infectious processes affecting the
myocardium may precipitate HF in patients with or without preexisting heart
disease
Infective endocarditis. The additional valvular damage, anemia,
fever, and myocarditis that often occur as a consequence of infective
endocarditis may, singly or in concert, frequently precipitate HF
Physical, dietary, fluid, environmental, and emotional
excesses.
The sudden augmentation of sodium intake as with a large meal, the
inappropriate discontinuation of pharmaceuticals to treat HF, blood
transfusions, physical overexertion, excessive environmental heat or humidity,
and emotional crises all may precipitate HF in patients with heart disease who
were previously compensated.
Systemic hypertension. Rapid elevation of arterial pressure, as may
occur in some instances of hypertension of renal origin or upon discontinuation
of antihypertensive medication in patients with essential hypertension, may
result in cardiac decompensation
Myocardial infarction. In patients with chronic but compensated
ischemic heart disease, a fresh infarct, sometimes otherwise silent clinically,
may further impair ventricular function and precipitate HF
Pulmonary embolism. Physically inactive patients with low cardiac
output are at increased risk of developing thrombi in the veins of the lower
extremities or the pelvis. Pulmonary emboli may result in further elevation of
pulmonary arterial pressure, which in turn may produce or intensify ventricular
failure. In the presence of pulmonary vascular congestion, such emboli also may
cause pulmonary infarction.
A
systematic search for these precipitating causes should be made in every
patient with the new development or recent intensification of HF. If properly
recognized, the precipitating cause of HF usually can be treated more
effectively than the underlying cause. Therefore, the prognosis in patients
with HF in whom a precipitating cause can be identified, treated, and
eliminated is more favorable than in patients in whom the underlying disease
process has progressed to the point of producing HF without a precipitating
cause.
Forms of Heart Failure
HF
may be described as systolic or diastolic, high-output or low-output,
acute or chronic, right-sided or left-sided, and forward
or backward. These descriptors are often useful in a clinical setting,
particularly early in the patient's course, but late in the course of chronic
HF the differences between them often become blurred.
Systolic Versus Diastolic Failure
The
distinction between these two forms of HF, described in Fig. 231-9, relates to
whether the principal abnormality is the inability of the ventricle to contract
normally and expel sufficient blood (systolic failure) or to relax and/or fill
normally (diastolic failure). The major clinical manifestations of systolic
failure relate to an inadequate cardiac output with weakness, fatigue, reduced
exercise tolerance, and other symptoms of hypoperfusion, while in diastolic HF
the manifestations relate principally to the elevation of filling pressures.
Many patients, particularly those who have both ventricular hypertrophy and
dilatation, exhibit abnormalities both of contraction and relaxation coexist.
Diastolic
HF may be caused by increased resistance to ventricular inflow and reduced
ventricular diastolic capacity (constrictive pericarditis and restrictive,
hypertensive, and hypertrophic cardiomyopathy), impaired ventricular relaxation
(acute myocardial ischemia), and myocardial fibrosis and infiltration
(restrictive cardiomyopathy).
High-Output versus Low-Output Heart Failure
It
is useful to classify patients with HF into those with a low cardiac output,
i.e., low-output HF, and those with an elevated cardiac output, i.e., high-output
HF. The former occurs secondary to ischemic heart disease, hypertension,
dilated cardiomyopathy, and valvular and pericardial disease, while the latter
is seen in patients with HF and hyperthyroidism, anemia, pregnancy,
arteriovenous fistulas, beriberi, and Paget's disease. In clinical practice,
however, low-output and high-output HF cannot always be readily distinguished.
The normal range of cardiac output is wide [2.2 to 3.5 (L/min)/m2];
in many patients with so-called low-output HF, the cardiac output may actually
be just within the normal range at rest (although lower than it had been
previously), but it fails to rise normally during exertion. On the other hand,
in patients with so-called high-output HF, the output may not exceed the upper
limits of normal (although it would have been elevated had it been measured
before HF supervened); rather, it may have fallen to within normal limits.
Regardless of the absolute level of the cardiac output, however, cardiac
failure may be said to be present when the characteristic clinical
manifestations described below are accompanied by a depression of the curve
relating ventricular end-diastolic volume to cardiac performance (see Fig.
231-6).
An
integral physiologic component of systolic HF is the delivery of an
inadequate quantity of oxygen required by the metabolizing tissues. In the
absence of peripheral shunting of blood, this is reflected in an abnormal widening
of the normal arterial-mixed venous oxygen difference (35 to 50 mL/L in the
basal state). In mild cases, such an abnormality may not be present at rest but
becomes evident only during exertion or other hypermetabolic states. In
patients with high cardiac output states, such as those associated with
arteriovenous fistula or thyrotoxicosis, the arterial-mixed venous oxygen
difference is normal or low. The mixed venous oxygen saturation is raised by
the admixture of blood that has been diverted away from the metabolizing
tissues, and it may be presumed that even in these patients the delivery of
oxygen to the latter is reduced despite the normal or even elevated mixed
venous oxygen saturation. When HF occurs in such patients, the arterial-mixed
venous oxygen difference, regardless of the absolute value, still exceeds the
level that existed prior to the development of HF. Therefore, the cardiac
output, though normal or even elevated, is lower than before HF supervened.
In
most forms of high-output HF, the heart is called on to pump abnormally large
quantities of blood in order to deliver the oxygen required by the metabolizing
tissues. The hemodynamic burden placed on the myocardium by the increased flow
load resembles that produced by chronic aortic regurgitation. In addition,
thyrotoxicosis and beriberi may also impair myocardial metabolism directly,
while very severe anemia may interfere with myocardial function by producing
myocardial anoxia, especially in the subendocardium and in the presence of
underlying obstructive coronary artery disease.
Acute versus Chronic Heart Failure
The
prototype of acute HF is the sudden development of a large myocardial
infarction or rupture of a cardiac valve in a patient who previously was
entirely well. Chronic HF is typically observed in patients with dilated
cardiomyopathy or multivalvular heart disease that develops or progresses
slowly. Acute HF is usually predominantly systolic, and the sudden reduction in
cardiac output often results in systemic hypotension without peripheral edema.
In contrast, in chronic HF, arterial pressure is ordinarily well maintained
until very late in the course, but there is often accumulation of edema.
Right-Sided versus Left-Sided Heart Failure
Many
of the clinical manifestations of HF result from the accumulation of excess
fluid behind either one or both ventricles (Chaps. 32 and 37). This fluid
usually localizes upstream to (behind) the ventricle that is initially
affected. For example, patients in whom the left ventricle is hemodynamically overloaded
(e.g., aortic stenosis) or weakened (e.g., postmyocardial infarction) develop
dyspnea and orthopnea as a result of pulmonary congestion, a condition referred
to as left-sided HF. In contrast, when the underlying abnormality
affects the right ventricle primarily (e.g., congenital valvular pulmonic
stenosis or pulmonary hypertension secondary to pulmonary thromboembolism),
symptoms resulting from pulmonary congestion are uncommon, and edema,
congestive hepatomegaly, and systemic venous distention, i.e., clinical
manifestations of right-sided HF, are more prominent. When HF has
existed for months or years, such localization of excess fluid behind the
failing ventricle may no longer exist. For example, patients with long-standing
aortic valve disease or systemic hypertension may develop ankle edema,
congestive hepatomegaly, and systemic venous distention late in the course of
their disease, even though the abnormal hemodynamic burden initially was placed
on the left ventricle. This occurs in part because of the secondary pulmonary
hypertension and resultant right-sided HF but also because of the retention of
salt and water characteristic of HF (Chap. 37). The muscle bundles composing
both ventricles are continuous, and both ventricles share a common wall, the
interventricular septum. Also, biochemical changes that occur in HF and that
may be involved in the impairment of myocardial function (Chap. 231), such as
norepinephrine depletion and alterations in the activity of myosin ATPase,
occur in the myocardium of both ventricles, regardless of the specific
chamber on which the abnormal hemodynamic burden is placed initially.
Backward versus Forward Heart Failure
For
many years a controversy has revolved around the question of the mechanism of
the clinical manifestations resulting from HF. The concept of backward HF
contends that in HF, one or the other ventricle fails to discharge its contents
or fails to fill normally. As a consequence, the pressures in the atrium and
venous system behind the failing ventricle rise, and retention of sodium and
water occurs as a consequence of the elevation of systemic venous and capillary
pressures and the resultant transudation of fluid into the interstitial space
(Chap. 37). In contrast, the proponents of the forward HF hypothesis
maintain that the clinical manifestations of HF result directly from an
inadequate discharge of blood into the arterial system. According to this
concept, salt and water retention is a consequence of diminished renal
perfusion and excessive proximal tubular sodium reabsorption and of excessive
distal tubular reabsorption through activation of the
renin-angiotensin-aldosterone (RAA) system.
A
rigid distinction between backward and forward HF (like a rigid
distinction between right and left HF) is artificial, since both mechanisms
appear to operate to varying extents in most patients with HF. However, the
rate of onset of HF often influences the clinical manifestations. For example,
when a large portion of the left ventricle is suddenly destroyed, as in
myocardial infarction, although stroke volume and blood pressure are suddenly
reduced (both manifestations of forward failure), the patient may succumb to
acute pulmonary edema, a manifestation of backward failure. If the patient
survives the acute insult, clinical manifestations resulting from a chronically
depressed cardiac output, including the abnormal retention of fluid within the
systemic vascular bed, may develop. Similarly, in the case of massive pulmonary
embolism, the right ventricle may dilate and the systemic venous pressure may
rise to high levels (backward failure), or the patient may develop shock
secondary to low cardiac output (forward failure), but this low-output state
may have to be maintained for some days before sodium and water retention
sufficient to produce peripheral edema occurs.
Redistribution of Cardiac Output
In
HF, systemic blood flow is redistributed so that the delivery of oxygen to
vital organs, such as the brain and myocardium, is maintained at normal or
near-normal levels, while flow to less critical areas, such as the cutaneous
and muscular beds and the viscera, is reduced. This redistribution serves as an
important compensatory mechanism when cardiac output is reduced. It is most
marked when a patient with HF exercises, but as HF advances, redistribution
occurs even in the basal state. Vasoconstriction mediated by the adrenergic
nervous system is largely responsible for redistribution, which in turn may be
responsible for many of the clinical manifestations of HF, such as fluid
accumulation (reduction of renal blood flow), low-grade fever (reduction of
cutaneous flow), and fatigue (reduction of muscle flow).
Salt and Water Retention
When
the volume of blood pumped by the left ventricle into the systemic vascular bed
is reduced, a complex sequence of adjustments occurs that ultimately results in
the abnormal accumulation of fluid. On the one hand, many of the troubling
clinical manifestations of HF are secondary to this excessive retention of
fluid; on the other, this abnormal fluid accumulation and the expansion of
blood volume that accompanies it also constitute an important compensatory
mechanism that tends to maintain cardiac output and therefore perfusion of the
vital organs. Except in the terminal stages of HF, the ventricle operates on an
ascending, albeit depressed and flattened, function curve (Fig. 231-6, p.
1313), and the augmented ventricular end-diastolic volume and pressure
characteristic of HF must be regarded as helping to maintain the reduced
cardiac output, despite causing pulmonary and/or systemic venous congestion.
Congestive
HF is also characterized by a complex series of neurohumoral adjustments. The
activation of the adrenergic nervous system is discussed on p. 1315; there is
also activation of the RAA system and increased release of antidiuretic hormone
and endothelin. These influences elevate systemic vascular resistance and
enhance sodium and water retention and potassium excretion. These actions are,
to a minor extent, opposed by the release of atrial and brain natriuretic
peptide, which also occurs in congestive HF. Patients with severe HF may
exhibit a reduced capacity to excrete a water load, which may result in
dilutional hyponatremia. In the presence of HF, effective filling of the
systemic arterial bed is reduced, a condition that initiates the renal and
hormonal changes mentioned above (Fig. 37-2).
The
elevation of systemic venous pressure and the alterations of renal and adrenal
function characteristic of HF vary in their relative importance in the
production of edema in different patients. The RAA axis is activated most
intensely by acute HF, and its activity tends to decline as HF becomes chronic.
In patients with tricuspid valve disease or constrictive pericarditis, the
elevated venous pressure and the transudation of fluid from systemic
capillaries appear to play the dominant role in edema formation. On the other
hand, severe edema may be present in patients with ischemic or hypertensive
heart disease, in whom systemic venous pressure is within normal limits or is
only minimally elevated. In such patients, the retention of salt and water is
probably due primarily to a redistribution of cardiac output and a concomitant
reduction in renal perfusion, as well as activation of the RAA axis. Regardless
of the mechanisms involved in fluid retention, untreated patients with chronic
congestive HF have elevations of total blood volume, interstitial fluid volume,
and body sodium. These abnormalities diminish after clinical compensation has
been achieved by effective treatment, especially with diuretics.
Clinical Manifestations of Heart Failure
Dyspnea
Respiratory
distress that occurs as the result of increased effort in breathing is the most
common symptom of HF (Chap. 32). In early HF, dyspnea is observed only during
activity, when it may simply represent an aggravation of the breathlessness
that occurs normally under these circumstances. As HF advances, however,
dyspnea appears with progressively less strenuous activity and ultimately is
present even when the patient is at rest. The principal difference between
exertional dyspnea in normal persons and in patients with HF is the degree of
activity necessary to induce this symptom. Cardiac dyspnea is observed most
frequently in patients with elevations of pulmonary venous and capillary
pressures. Such patients usually have engorged pulmonary vessels and
interstitial pulmonary edema, which may be evident on radiologic examination.
This interstitial pulmonary edema reduces the compliance of the lungs and thereby
increases the work of the respiratory muscles required to inflate the lungs.
The activation of receptors in the lungs results in the rapid, shallow
breathing characteristic of cardiac dyspnea. The oxygen cost of breathing is
increased by the excessive work of the respiratory muscles. This is coupled
with the diminished delivery of oxygen to these muscles, which occurs as a
consequence of the reduced cardiac output and which may contribute to fatigue
of the respiratory muscles and the sensation of shortness of breath.
Orthopnea
Dyspnea
in the recumbent position is usually a later manifestation of HF than
exertional dyspnea. Orthopnea occurs because of the redistribution of fluid
from the abdomen and lower extremities into the chest during recumbency causing
an increase in the pulmonary capillary hydrostatic pressure, as well as
elevation of the diaphragm accompanying supine posture. Patients with orthopnea
must elevate their heads on several pillows at night and frequently awaken
short of breath or coughing (the so-called nocturnal cough) if their heads slip
off the pillows. The sensation of breathlessness is usually relieved by sitting
upright, since this position reduces venous return and pulmonary capillary
pressure, and many patients report that they find relief from sitting in front
of an open window. In advanced HF, orthopnea may become so severe that patients
cannot lie down at all and must spend the entire night in a sitting position.
On the other hand, in other patients with long-standing, severe left
ventricular failure, symptoms of pulmonary congestion may actually diminish
with time as the function of the right ventricle becomes impaired.
Paroxysmal (Nocturnal) Dyspnea
This
term refers to attacks of severe shortness of breath and coughing that generally
occur at night, usually awaken the patient from sleep, and may be quite
frightening. Though simple orthopnea may be relieved by sitting upright at the
side of the bed with legs dependent, in the patient with paroxysmal nocturnal
dyspnea, coughing and wheezing often persist even in this position. Paroxysmal
nocturnal dyspnea may be caused in part by the depression of the respiratory
center during sleep, which may reduce ventilation sufficiently to lower
arterial oxygen tension, particularly in patients with interstitial lung edema
and reduced pulmonary compliance. Also, ventricular function may be further
impaired at night because of reduced adrenergic stimulation of myocardial
function. Cardiac asthma is closely related to paroxysmal nocturnal
dyspnea and nocturnal cough and is characterized by wheezing secondary to
bronchospasm-most prominent at night. Acute pulmonary edema (Chap. 32)
is a severe form of cardiac asthma due to marked elevation of pulmonary
capillary pressure leading to alveolar edema, associated with extreme shortness
of breath, rales over the lung fields, and the transudation and expectoration
of blood-tinged fluid. If not treated promptly, acute pulmonary edema may be
fatal.
Cheyne-Stokes Respiration
Also
known as periodic respiration or cyclic respiration,
Cheyne-Stokes respiration is characterized by diminished sensitivity of the
respiratory center to arterial PCO2. There is an apneic phase,
during which the arterial PO2 falls and the arterial PCO2
rises. These changes in the arterial blood stimulate the depressed respiratory
center, resulting in hyperventilation and hypocapnia, followed in turn by
recurrence of apnea. Cheyne-Stokes respiration occurs most often in patients
with cerebral atherosclerosis and other cerebral lesions, but the prolongation
of the circulation time from the lung to the brain that occurs in HF,
particularly in patients with hypertension and coronary artery disease and
associated cerebral vascular disease, also appears to precipitate this form of
breathing.
Fatigue and Weakness
These
nonspecific but common symptoms of HF are related to the reduction of perfusion
of skeletal muscle. Exercise capacity is reduced by the limited ability of the
failing heart to increase its output and deliver oxygen to the exercising muscle.
Abdominal Symptoms
Anorexia
and nausea associated with abdominal pain and fullness are frequent complaints
and may be related to the congested liver and portal venous system.
Cerebral Symptoms
In
severe HF, particularly in elderly patients with accompanying cerebral
arteriosclerosis, reduced cerebral perfusion, and arterial hypoxemia, there may
be alterations in the mental state characterized by confusion, difficulty in
concentration, impairment of memory, headache, insomnia, and anxiety. Nocturia
is common in HF and may contribute to insomnia.
Physical Findings
In
moderate HF, the patient is in no distress at rest except that he or she may be
uncomfortable when lying flat for more than a few minutes. In more severe HF,
the pulse pressure may be diminished, reflecting a reduction in stroke volume,
and the diastolic arterial pressure may be elevated as a consequence of
generalized vasoconstriction. In acute HF, severe hypotension may be present.
There may be cyanosis of the lips and nail beds (Chap. 36) and sinus
tachycardia, and the patient may insist on sitting upright. Systemic venous
pressure is often abnormally elevated in HF, and this may be reflected in
distention of the jugular veins. In the early stages of HF, the venous pressure
may be normal at rest but may become abnormally elevated during and immediately
after exertion as well as with sustained pressure on the abdomen (positive
abdominojugular reflux).
Third
and fourth heart sounds are often audible but are not specific for HF, and pulsus
alternans, i.e., a regular rhythm in which there is alternation of strong
and weak cardiac contractions and therefore alternation in the strength of the
peripheral pulses, may be present. Pulsus alternans, a sign of severe HF, may
be detected by sphygmomanometry and in more severe instances by palpation; it
frequently follows an extrasystole and is observed most commonly in patients
with cardiomyopathy or hypertensive or ischemic heart disease.
Pulmonary Rales
Moist,
inspiratory, crepitant rales and dullness to percussion over the lung bases are
common in patients with HF and elevated pulmonary venous and capillary
pressures. In patients with pulmonary edema, rales may be heard widely over
both lung fields; they are frequently coarse and sibilant and may be accompanied
by expiratory wheezing. Rales may, however, be caused by many conditions other
than left ventricular failure. Some patients with long-standing HF have no
rales because of increased lymphatic drainage of alveolar fluid.
Cardiac Edema
This
is usually symmetric and dependent, occurring in the legs, particularly in the
pretibial region and ankles in ambulatory patients, in whom it is most
prominent in the evening. Cardiac edema occurs in the sacral region of patients
who are bed-ridden. Pitting edema of the arms and face occurs rarely and then
only late in the course of HF.
Hydrothorax and Ascites
Pleural
effusion in congestive HF results from the elevation of pleural capillary
pressure and transudation of fluid into the pleural cavities. Since the pleural
veins drain into both the systemic and pulmonary veins, hydrothorax
occurs most commonly with marked elevation of pressure in both venous systems
but also may be seen with marked elevation of pressure in either venous bed. It
is more frequent in the right pleural cavity than in the left. Ascites
also occurs as a consequence of transudation and results from increased
pressure in the hepatic veins and the veins draining the peritoneum (Chap. 46).
Marked ascites occurs most frequently in patients with tricuspid valve disease
and constrictive pericarditis.
Congestive Hepatomegaly
An
enlarged, tender, pulsating liver also accompanies systemic venous hypertension
and is observed not only in the same conditions in which ascites occurs but
also in milder forms of HF from any cause. With prolonged, severe hepatomegaly,
as in patients with tricuspid valve disease or chronic constrictive
pericarditis, enlargement of the spleen, i.e., congestive splenomegaly, may
also occur.
Jaundice
This
is a late finding in HF and is associated with elevations of both the direct-
and indirect-reacting bilirubin; it results from impairment of hepatic function
secondary to hepatic congestion and the hepatocellular hypoxia associated with
central lobular atrophy. Hepatic enzymes are frequently elevated. If hepatic
congestion occurs acutely, the jaundice may be severe and the enzymes
strikingly elevated.
Cardiac Cachexia
With
severe chronic HF there may be serious weight loss and cachexia because of (1)
elevation of circulating concentrations of tumor necrosis factor; (2) elevation
of the metabolic rate, which results in part from the extra work performed by
the respiratory muscles, the increased oxygen needs of the hypertrophied heart,
and/or the discomfort associated with severe HF; (3) anorexia, nausea, and
vomiting due to central causes, to digitalis intoxication, or to congestive
hepatomegaly and abdominal fullness; (4) impairment of intestinal absorption
due to congestion of the intestinal veins; and (5) rarely, due to protein-losing
enteropathy in patients with particularly severe failure of the right side of
the heart.
Other Manifestations
With
reduction of blood flow, the extremities may be cold, pale, and diaphoretic.
Urine flow is depressed, and the urine contains albumin and has a high specific
gravity and a low concentration of sodium. In addition, prerenal azotemia may
be present. In patients with long-standing severe HF, impotence and depression
are common.
Roentgenographic and Echocardiographic Findings
In
addition to the enlargement of the particular chambers characteristic of the
lesion responsible for HF, distention of pulmonary veins and redistribution to
the apices is common in patients with HF and elevated pulmonary vascular
pressures. Also, pleural effusions may be evident and associated with
interlobar effusions.
Differential Diagnosis
The diagnosis of congestive HF may be established by observing some
combination of the clinical manifestations of HF described above, together with
the findings characteristic of one of the etiologic forms of heart disease.
Table 232-1 shows the Framingham criteria, which are useful in the diagnosis of
HF. Since chronic HF is often associated with cardiac enlargement, the
diagnosis should be questioned, but is by no means excluded, when all chambers
are normal in size. Two-dimensional echocardiography (Chap. 227) is
particularly useful in assessing the dimensions of each cardiac chamber. HF is
sometimes difficult to distinguish from pulmonary disease, and the differential
diagnosis is discussed in Chap. 32. Pulmonary embolism also presents many of
the manifestations of HF, but hemoptysis, pleuritic chest pain, a right
ventricular lift, and the characteristic mismatch between ventilation and
perfusion on lung scan should point to this diagnosis.
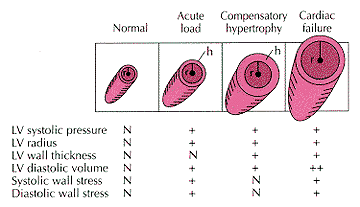
Figure 232-3: Chest roentgenogram of patient with heart
failure. This roentgenogram demonstrates cardiomegaly (cardiothoracic ratio
0.77), pulmonary congestion, and bilateral pleural effusions (note blunted
costophrenic angles). [JB Young: Assessment of heart failure, in WS Colucci:
Heart failure: Cardiac Function and Dysfunction, in E Braunwald (series ed):
Atlas of Heart Diseases, vol 4. Philadelphia, Current Medicine, 1995, p 7.16.]
Table 232-1:
|
|||||||||||||||||||
|
aTo establish a clinical diagnosis of congestive
heart failure by these criteria, at least one major and two minor criteria
are required. |
Ankle
edema may be due to varicose veins, cyclic edema, or gravitational effects
(Chap. 37), but in these patients there is no jugular venous hypertension at
rest or with pressure over the abdomen. Edema secondary to renal disease can
usually be recognized by appropriate renal function tests and urinalysis and is
rarely associated with elevation of venous pressure. Enlargement of the liver
and ascites occur in patients with hepatic cirrhosis and also may be
distinguished from HF by normal jugular venous pressure and absence of a
positive abdominojugular reflux.
![]() Treatment
Treatment
(See
Practice Guidelines, Table 232-4) The treatment of HF may be divided into four
components: (1) removal of the precipitating cause, (2) correction of the
underlying cause, (3) prevention of deterioration of cardiac function, and (4)
control of the congestive HF state. The first two components are discussed in
other chapters together with each specific disease entity or complication.
Examples of removal of precipitating causes are the treatment of pneumococcal
pneumonia or the restoration of sinus rhythm in a patient with atrial
fibrillation. In many instances, surgical treatment will correct or at least
alleviate the underlying cause of HF. The third component of the treatment of
HF, i.e., the prevention of deterioration of cardiac function, involves the
administration of angiotensin-converting enzyme (ACE) inhibitors and ![]() -adrenergic
blockers as well as reduction of cardiac work load. Control of the congestive
heart failure state requires reduction of the excessive retention of salt and
water as well as enhancement of myocardial contractility. The vigor with which
each of these measures is pursued in any individual patient should depend on
the severity of HF and the tempo of the disease. Following effective treatment,
recurrence of the clinical manifestations of HF can often be prevented by
continuing those measures that were originally effective.
-adrenergic
blockers as well as reduction of cardiac work load. Control of the congestive
heart failure state requires reduction of the excessive retention of salt and
water as well as enhancement of myocardial contractility. The vigor with which
each of these measures is pursued in any individual patient should depend on
the severity of HF and the tempo of the disease. Following effective treatment,
recurrence of the clinical manifestations of HF can often be prevented by
continuing those measures that were originally effective.
Table 232-4: Practice Guidelines for Heart
Failure
|
While
a simple rule for the treatment of all patients with HF cannot be formulated
because of its varied etiologies, hemodynamic features, clinical
manifestations, and severity of HF, insofar as the treatment of chronic
congestive failure is concerned, the administration of an ACE inhibitor retards
the development of HF and should be begun early in patients with left
ventricular systolic dysfunction (ejection fraction < 0.40), even if they
are asymptomatic. Then, as symptoms develop, simple measures such as moderate
restriction of activity and sodium intake and oral diuretics should be tried. ![]() -adrenergic
receptor blockers and digitalis glycosides are given for patients with systolic
HF. If these measures are insufficient, the next step is more rigorous
restriction of salt intake and higher doses and multiple diuretics. If HF persists,
hospitalization with rigid salt restriction, bed rest, intravenous
vasodilators, and positive inotropic agents are tried. Assisted circulation and
cardiac transplantation (Chap. 233) are considered for patients with severe,
intractable HF and a poor prognosis.
-adrenergic
receptor blockers and digitalis glycosides are given for patients with systolic
HF. If these measures are insufficient, the next step is more rigorous
restriction of salt intake and higher doses and multiple diuretics. If HF persists,
hospitalization with rigid salt restriction, bed rest, intravenous
vasodilators, and positive inotropic agents are tried. Assisted circulation and
cardiac transplantation (Chap. 233) are considered for patients with severe,
intractable HF and a poor prognosis.
Prevention of Deterioration of Myocardial
Infarction
Chronic
activation of the RAA axis and of the sympathetic nervous systems in HF result
in a maladaptive response and cause further deterioration of cardiac function
and/or potentially fatal arrhythmias (Chap. 231). Drugs that block these two
systems have been found to be useful in the management of HF (Tables 232-2 and
232-3).
Table 232-2: Overview of Angiotensin-Converting
Enzyme Inhibitors in Heart Failure
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 232-3: Clinical Trials in Heart Failure
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Angiotensin-Converting
Enzyme Inhibitors
In
many patients with HF, the left ventricular afterload is increased as a
consequence of the several neural and humoral influences that act to constrict
the peripheral vascular bed. In addition to the vasoconstriction, the
ventricular end-diastolic and -systolic volumes rise in systolic HF. As a
consequence of the operation of Laplace's law, which relates myocardial wall
tension to the product of intraventricular pressure and radius (both of which
may become elevated in HF), the aortic impedance, i.e., the force that opposes
left ventricular ejection, or the ventricular afterload, rises, which reduces
stroke volume (Fig. 231-10, p. 1316). In many patients with systolic HF, a
modest reduction of systemic vascular resistance and afterload elevates the
stroke volume and reduces the elevated ventricular filling pressure of the
failing ventricle.
The
pharmacologic reduction of impedance to left ventricular ejection with an ACE
inhibitor represents an important component of the management of HF. This approach
may be particularly helpful in (but is by no means limited to) patients with
systolic HF due to myocardial infarction (Chap. 243), and in patients with
valvular regurgitation (Chap. 236). ACE inhibition should not be used in
hypotensive patients. In patients with both acute and chronic systolic HF who
are treated with ACE inhibitors, cardiac output rises, the pulmonary wedge
pressure falls, the signs and symptoms of HF are relieved, and a new steady
state is achieved in which cardiac output is higher and afterload lower with no
or only mild reduction of arterial pressure. The administration of ACE
inhibitors has been shown to prevent or retard the development of HF in
patients with left ventricular dysfunction without HF, to reduce symptoms,
enhance exercise performance, and to reduce long-term mortality when they are
begun in such patients shortly after acute myocardial infarction. These
beneficial effects are related only in part to the salutary hemodynamic
effects, i.e., the reduction of preload and afterload. Their major effect
appears to be on inhibition of local (tissue) renin-angiotensin systems.
Lisinopril
in doses of 20 mg qd or enalapril 10 mg bid have been shown to be useful in the
management of heart failure.
Angiotensin Receptor Blockers
In
patients who cannot tolerate ACE inhibitors (because of cough, angioneurotic
edema, leukopenia), an angiotensin II receptor blocker (type AT1) antagonist
(e.g., losartan 50 mg qid) may be used instead.
Aldosterone Antagonist
The
activation of the RAA axis in HF increases not only circulating and tissue
angiotensin II but also aldosterone. The latter, in addition to causing sodium
retention and worsening edema (Chaps. 331 and 37), causes sympathetic
activation, myocardial, vascular, and perivascular fibrosis and reduces
arterial compliance. In one large multicenter trial in patients with advanced
heart failure and reduced ejection fraction (RALES), spironolactone, 25 mg/d
reduced total mortality, as well as sudden death and death from pump failure
(Table 232-3). Since spironolactone is also a useful diuretic (see below), its
widespread use in systolic heart failure should be considered.
![]() -Adrenoceptor
Blockers
-Adrenoceptor
Blockers
While
the abrupt administration of large doses of ![]() -adrenergic
receptor blockers can intensify HF, the administration of gradually escalating
doses of metoprolol, carvedilol, and bisoprolol have been reported to improve
the symptoms of HF, and to reduce all-cause death, cardiovascular death, sudden
death, and pump failure death (Table 232-3). In patients with moderately severe
HF (classes II and III), the administration of 12.5 mg metoprolol CR/XL qd,
increasing over 4 weeks to a target dose of 200 mg qid, has been shown to be
beneficial.
-adrenergic
receptor blockers can intensify HF, the administration of gradually escalating
doses of metoprolol, carvedilol, and bisoprolol have been reported to improve
the symptoms of HF, and to reduce all-cause death, cardiovascular death, sudden
death, and pump failure death (Table 232-3). In patients with moderately severe
HF (classes II and III), the administration of 12.5 mg metoprolol CR/XL qd,
increasing over 4 weeks to a target dose of 200 mg qid, has been shown to be
beneficial. ![]() -Adrenoceptor
blockers are not indicated in HF patients who are unstable, in New York Heart
Association Class IV, in HF patients shortly after acute myocardial infarction,
or in those with HF and normal ejection fraction, i.e., with diastolic HF.
-Adrenoceptor
blockers are not indicated in HF patients who are unstable, in New York Heart
Association Class IV, in HF patients shortly after acute myocardial infarction,
or in those with HF and normal ejection fraction, i.e., with diastolic HF.
Reduction of Cardiac Work Load
This
consists of reducing physical activity, instituting emotional rest, and
reducing afterload (see above). Modest restriction of physical activity in mild
cases and rest in bed or in a chair in severe failure are useful. In acute,
severe failure, meals should be small in quantity, but more frequent, and every
effort should be made to diminish the patient's anxiety; sometimes drugs such
as diazepam (2 to 5 mg tid) for several days are useful. Physical and emotional
rest tends to lower arterial pressure and reduce the load on the myocardium by
diminishing the requirements for cardiac output.
Reduced
physical exertion should be continued for several days after the patient's
condition has stabilized. The hazards of phlebothrombosis and pulmonary
embolism which occur with bed rest may be reduced with anticoagulants, leg
exercises, and elastic stockings. Absolute bed rest is rarely required
or advisable, and the patient should ordinarily be encouraged to sit in a
chair. Heavy sedation should be avoided. In ambulatory patients with chronic,
moderately severe HF, additional periods of rest on weekends frequently allow
continuation of gainful employment. Following recovery from HF, the patient's
activities should be assessed, and often, professional, community, and/or
family responsibilities should be curtailed. Intermittent rest during the day
(e.g., a scheduled 1-h nap or rest following lunch) and the avoidance of
strenuous exertion are often helpful. Regular, nonexhausting exercise such as
walking or riding a stationary-bicycle ergometer as tolerated should be
employed once the patient has become compensated. Weight reduction by
restriction of caloric intake in obese patients with HF also diminishes cardiac
work load and is an essential component of the therapeutic program.
Control of Excessive Fluid
Many
of the clinical manifestations of HF result from expansion of the extracellular
fluid volume. A negative sodium balance can be achieved by reducing the dietary
intake and increasing the urinary excretion of this ion with the aid of
diuretics. Rarely, in severe HF, mechanical removal of extracellular fluid by
means of thoracentesis and paracentesis may be necessary.
Diet
In
patients with mild HF, symptomatic improvement may result simply from reducing
the sodium intake, particularly if accompanied by periods of physical rest. The
normal diet contains approximately 6 to 10 g sodium chloride; this intake can
be reduced by half simply by excluding salt-rich foods and salt added at the
table. Reduction of the ordinary dietary intake to approximately one-fourth of
normal may be achieved if, in addition, all salt is omitted from cooking. In
patients with severe HF who have fluid accumulation despite diuretic therapy
(see below), the dietary intake of sodium chloride should be reduced to between
500 and 1000 mg, and in order to achieve this, milk, cheese, bread, cereals,
canned vegetables and soups, some salted cuts of meat, and some fresh
vegetables (including spinach, celery, and beets) must be eliminated. A variety
of fresh fruit, green vegetables, specially processed breads and milk, and salt
substitutes are permissible. Late in the course of HF, dilutional hyponatremia
may develop in patients who are unable to excrete a water load, sometimes
because of excessive secretion of antidiuretic hormone. In such cases, water
intake as well as sodium intake must be restricted.
Calories
should be restricted in obese patients with HF. In patients with severe HF and
cardiac cachexia, on the other hand, an attempt must be made to maintain
nutritional intake and to avoid caloric and vitamin deficiencies; nutritional
supplements may be in order.
Diuretics
Diuretics
should be given to relieve fluid accumulation and thus reduce or prevent edema
and jugular venous distention. A variety of diuretic agents are available
(Table 246-6, p. 1422), and almost all are effective in patients with mild HF.
However, in the more severe forms of HF, the selection of diuretics is more
difficult, and abnormalities in serum electrolytes must be taken into account.
Overtreatment must be avoided, since the resultant hypovolemia may reduce
cardiac output, interfere with renal function, and produce profound weakness
and lethargy.
Thiazide Diuretics
These
agents are used widely and are useful by themselves in patients with mild HF
and in combination with other diuretics in those with severe HF. In patients
with chronic mild or moderate HF, the continued administration of a thiazide
diuretic abolishes or diminishes the need for rigid dietary sodium restriction,
although salty foods and table salt still should be avoided. Thiazide diuretics
reduce the reabsorption of sodium and chloride in the first half of the distal
convoluted tubule and a portion of the cortical ascending limb of the loop of
Henle, and water follows the unreabsorbed salt. Thiazides fail to increase free
water clearance and in some instances reduce it. This may result in the
excretion of a hypertonic urine and may contribute to dilutional hyponatremia.
As a consequence of increased delivery of sodium to the distal nephron,
sodium-potassium ion exchange is enhanced, and kaliuresis results. In contrast
to the loop diuretics, which enhance calcium excretion, the thiazides have the
opposite effect.
Thiazide
diuretics are effective and useful in the treatment of HF as long as the
glomerular filtration rate exceeds approximately 50% of normal. Chlorothiazide
is administered in doses of up to 500 mg every 6 h. Many derivatives of this
compound are available but differ principally in dosage and duration of action.
Chlorthalidone (25 to 50 mg/d) is especially useful since it may be
administered once daily.
Potassium
depletion and metabolic alkalosis (the latter due to increased H+
secretion as a substitute for the depleted intracellular stores of potassium)
are the chief adverse metabolic effects following prolonged administration of
the thiazides, of metolazone, and of the loop diuretics. Hypokalemia may seriously
enhance the dangers of digitalis intoxication (see below), and induce fatigue
and lethargy; these may be prevented by oral supplementation with potassium
chloride or preferably by the addition of a potassium-retaining diuretic, such
as a spironolactone or triamterene. Other side effects of thiazides include
reduction of the excretion of uric acid, which may lead to hyperuricemia, and
impaired glucose tolerance, which rarely may precipitate hyperosmolar coma in
poorly regulated diabetic patients. Skin rashes, thrombocytopenia, and
granulocytopenia have also been reported.
Metolazone
This
quinethazone derivative has a site of action and potency similar to those of
the thiazides but has been reported to be effective in the presence of moderate
renal failure. The usual dose is 5 to 10 mg/d. Metolazone may be added to
thiazide and loop diuretics in severe HF.
Furosemide, Bumetanide, Ethacrynic Acid,
Piretanide, and Torsemide
These
"loop" diuretics are similar physiologically but differ chemically
from one another. These drugs reversibly inhibit the reabsorption of sodium,
potassium, and chloride in the thick ascending limb of Henle's loop, apparently
by blocking a cotransport system in the luminal membrane. They may induce renal
cortical vasodilatation and can produce rates of urine formation that may be as
high as one-fourth of the glomerular filtration rate. Metabolic alkalosis may
be caused by a large increase in the urinary excretion of chloride, hydrogen,
and potassium ions. Hypokalemia, hyperuricemia, and hyperglycemia are observed
occasionally, as with thiazide diuretics. The reabsorption of free water is
decreased. All five of these drugs are readily absorbed orally, are excreted in
the bile and urine, and are usually effective both intravenously and by mouth.
Weakness, nausea, and dizziness may complicate the administration of all loop
diuretics; ethacrynic acid has been associated with transient or even permanent
deafness as well as with skin rash and granulocytopenia.
These
powerful diuretics are useful in all forms of HF, particularly in patients with
otherwise refractory HF and pulmonary edema. They have been shown to be
effective in patients with hypoalbuminemia, hyponatremia, hypochloremia,
hypokalemia, and with reductions in the glomerular filtration rate and to
produce a diuresis in patients in whom thiazide diuretics and aldosterone
antagonists, alone and in combination, are ineffective. In patients with
refractory HF, the action of loop diuretics may be potentiated by intravenous
administration and by the addition of other diuretics, i.e., thiazides,
metozalone, osmotic diuretics, and the potassium-sparing diuretics-spironolactone,
triamterene, and amiloride.
Aldosterone Antagonists
These
agents act on the cortical collecting ducts, are relatively weak, and therefore
are rarely indicated as sole agents. However, their potassium-sparing
properties make them particularly useful in conjunction with the more potent
kaliuretic agents, i.e., the loop diuretics, thiazides, and metozalone. The
potassium-sparing agents fall into two classes.
The
spironolactones resemble aldosterone structurally and act by competitive
inhibition of aldosterone, thereby blocking the exchange between sodium and
both potassium and hydrogen in the distal tubules and collecting ducts. These
agents produce a sodium diuresis, and in contrast to the thiazides, ethacrynic
acid, and furosemide, they result in potassium retention. Although secondary
hyperaldosteronism exists in some patients with congestive HF, the
spironolactones are effective even in patients in whom the serum aldosterone
concentration is within normal limits.
Spironolactone
may be administered in doses of 25 mg daily to 50 mg three to four times daily
by mouth. The maximal effect of this regimen is not observed for approximately
4 days. Spironolactones are most effective when administered in combination
with loop and/or thiazide diuretics. The opposing action of these drugs on
urine and serum potassium makes possible a sodium diuresis without either
hyper- or hypokalemia when spironolactone and one of these other agents are
administered in combination. Also, since spironolactone, triamterene, and
amiloride act on the distal tubule, they are particularly effective when used
in combination with one of these other diuretics that act more proximally.
Spironolactone, triamterene, and amiloride should not be administered alone to
patients with hyperkalemia, renal failure, or hyponatremia. Reported
complications of Aldactone A include nausea, epigastric distress, mental confusion,
drowsiness, gynecomastia, and erythematous eruptions.
As
mentioned above, a lower dose of spironolactone (25 mg/d), which exerts little
if any diuretic effect, has been shown to prolong life in patients with
advanced HF (Table 232-3).
Triamterene and amiloride exert renal effects
similar to those of the spironolactones; i.e., they block sodium reabsorption
and secondarily inhibit potassium secretion in the distal tubules. However,
their action does not depend on the presence of aldosterone. The effective dose
of triamterene is 100 mg once or twice daily, and that of amiloride is 5 mg
daily. Side effects include nausea, vomiting, diarrhea, headache,
granulocytopenia, eosinophilia, and skin rash. Both triamterene and the
chemically unrelated diuretic amiloride resemble Aldactone A in that their
diuretic potency is not great, but they are effective in preventing the
hypokalemia characteristic of loop diuretics and thiazides. A number of
diuretic preparations contain a combination of a thiazide and either
triamterene or amiloride in a single capsule. They may be useful in patients
who develop hypokalemia with a thiazide but should not be used in patients with
impaired renal function and/or hyperkalemia.
When
choosing a diuretic, orally administered loop diuretics or thiazides are
the agents of choice in the treatment of chronic cardiac edema of mild to
moderate degree in patients without hyperglycemia, hyperuricemia, or
hypokalemia. Spironolactones, triamterene, and amiloride are not potent
diuretics when used alone, but they potentiate the thiazide and loop diuretics.
Loop diuretics, given alone or with spironolactone or triamterene, are the
agents of choice in patients with severe HF refractory to other diuretics. In
very severe HF, the combination of a loop diuretic, a thiazide, and a
potassium-sparing diuretic is required.
Vasodilators
Direct
vasodilators may be useful in patients with severe, acute HF who demonstrate
systemic vasoconstriction despite ACE inhibitor therapy. The ideal vasodilator
for the treatment of acute HF should have a rapid onset and brief
duration of action when administered by intravenous infusion; sodium
nitroprusside (0.1 to 3.0 ![]() g/kg
per minute) qualifies as such a drug, but its use requires careful monitoring
of the arterial pressure and, if possible, of the pulmonary artery wedge
pressure. The combination of hydralazine (up to 300 mg qd orally) and
isosorbide diuretics (up to 160 mg qd orally) may be useful for chronic oral
administration.
g/kg
per minute) qualifies as such a drug, but its use requires careful monitoring
of the arterial pressure and, if possible, of the pulmonary artery wedge
pressure. The combination of hydralazine (up to 300 mg qd orally) and
isosorbide diuretics (up to 160 mg qd orally) may be useful for chronic oral
administration.
Enhancement of Myocardial Contractility
Digitalis
The
improvement of myocardial contractility by means of cardiac glycosides is
useful in the control of HF. Digoxin, which has a half-life of 1.6 days,
is filtered in the glomeruli and secreted by the renal tubules. Significant
reductions of the glomerular filtration rate reduce the elimination of digoxin
and, therefore, may prolong digoxin's effect, allowing it to accumulate to
toxic levels. In patients with normal renal function, a plateau concentration
in the blood and tissue is reached after 5 days of daily maintenance treatment
without a loading dose (see Fig. 70-2).
Mechanism of Action
The
most important effect of digitalis on cardiac muscle is to shift its
force-velocity relation upward (Fig. 231-5, p. 1313). Cardiac glycosides
inhibit the monovalent cation transport enzyme-coupled Na+,K+-ATPase
and increase intracellular sodium content; this, in turn, increases
intracellular Ca2+ through a Na+-Ca2+ exchange
carrier mechanism. The increased myocardial uptake of Ca2+ augments
Ca2+ released to the myofilaments during excitation and, therefore,
invokes a positive inotropic response.
Cardiac
glycosides also produce alterations in the electrical properties of both the
contractile cells and the specialized automatic cells, leading to increased
automaticity and ectopic impulse activity. They also prolong the effective refractory
period of the atrioventricular node and thereby slow ventricular rate in
atrial flutter and fibrillation.
Use in Heart Failure
Digitalis
is particularly effective in patients with systolic HF complicated by atrial
flutter and fibrillation and a rapid ventricular rate, who benefit from both slowing
of the ventricular rate and the positive inotropic effect. Although digitalis
does not improve survival in patients with systolic HF and sinus rhythm, it
reduces the need for hospitalization. By stimulating myocardial contractility
moderately, digitalis improves ventricular emptying; i.e., it increases cardiac
output, augments the ejection fraction, promotes diuresis, and reduces the
elevated diastolic pressure and volume and the end-systolic volume of the
failing ventricle. This action reduces symptoms resulting from pulmonary
vascular congestion and elevated systemic venous pressure. Digitalis is of
little or no value in patients with HF, sinus rhythm, and the following
conditions: hypertrophic cardiomyopathy, myocarditis, mitral stenosis, chronic constrictive
pericarditis, and any form of diastolic HF.
The
maintenance dose of digoxin is 0.25 mg qd for most adults; in the elderly and
others with mild impairment of renal function, it is 0.125 mg qd. Loading
doses, four times the maintenance dose, may be administered in acute systolic
failure.
Digitalis Intoxication
This
is a serious and potentially fatal complication. Advanced age, acute myocardial
infarction or ischemia, hypoxemia, magnesium depletion, renal insufficiency,
hypercalcemia, electrical cardioversion, and hypothyroidism all may reduce
tolerance to digitalis. The most common precipitating cause of digitalis
intoxication, however, is depletion of potassium stores, which often occurs in
patients with HF as a result of diuretic therapy and secondary
hyperaldosteronism.
Anorexia,
nausea, and vomiting are among the earliest signs of digitalis intoxication.
The most frequent disturbances of cardiac rhythm are ventricular premature
beats, bigeminy, ventricular tachycardia, and, rarely, ventricular fibrillation.
Atrioventricular block of varying degrees of severity may occur. Nonparoxysmal
atrial tachycardia with variable atrioventricular block is characteristic of
digitalis intoxication. Chronic digitalis intoxication may be insidious in
onset and characterized by exacerbations of HF, weight loss, cachexia,
neuralgias, gynecomastia, yellow vision, and delirium.
The
administration of quinidine, verapamil, amiodarone, and propafenone to patients
receiving digoxin raises the serum concentration of the latter by reducing both
the renal and nonrenal elimination of digoxin and by reducing its volume of
distribution. These drugs increase the propensity to digitalis intoxication,
and the dose of digitalis should be reduced by half in patients receiving these
drugs.
Treatment of Digitalis Intoxication
When
tachyarrhythmias result from digitalis intoxication, withdrawal of the drug and
treatment with ![]() -adrenoceptor
blocker or lidocaine are indicated. If hypokalemia is present, potassium should
be administered cautiously and by the oral route. Fab fragments of purified,
intact digitalis antibodies are a potentially lifesaving approach to the
treatment of severe intoxication.
-adrenoceptor
blocker or lidocaine are indicated. If hypokalemia is present, potassium should
be administered cautiously and by the oral route. Fab fragments of purified,
intact digitalis antibodies are a potentially lifesaving approach to the
treatment of severe intoxication.
Sympathomimetic Amines
Two
sympathomimetic amines that act largely on ![]() -adrenergic
receptors-dopamine and dobutamine-improve myocardial contractility (Table 72-1)
and are effective in the management of HF; they must be administered by
constant intravenous infusion for up to 1 week and are useful in patients with
intractable, severe HF, particularly those with a reversible component, such as
exists in patients who have undergone cardiac surgery, in patients with acute
myocardial infarction and shock or pulmonary edema, and in patients with
refractory HF as a "bridge" to transplantation. While these
sympathomimetic amines improve the hemodynamics and symptoms in these
conditions, it is not clear that they improve survival. Their administration
should be accompanied by careful and continuous monitoring of the
electrocardiogram, arterial pressure, and, if possible, pulmonary artery wedge
pressure.
-adrenergic
receptors-dopamine and dobutamine-improve myocardial contractility (Table 72-1)
and are effective in the management of HF; they must be administered by
constant intravenous infusion for up to 1 week and are useful in patients with
intractable, severe HF, particularly those with a reversible component, such as
exists in patients who have undergone cardiac surgery, in patients with acute
myocardial infarction and shock or pulmonary edema, and in patients with
refractory HF as a "bridge" to transplantation. While these
sympathomimetic amines improve the hemodynamics and symptoms in these
conditions, it is not clear that they improve survival. Their administration
should be accompanied by careful and continuous monitoring of the
electrocardiogram, arterial pressure, and, if possible, pulmonary artery wedge
pressure.
Dopamine is a naturally occurring immediate precursor of
norepinephrine and has a combination of actions that makes it particularly
useful in the treatment of a variety of hypotensive states of HF. At very low
doses, i.e., 1 to 2 (![]() g/kg)/min,
it dilates renal and mesenteric blood vessels through stimulation of specific
dopaminergic receptors, thereby augmenting renal and mesenteric blood flow and
sodium excretion. In the range of 2 to 10 (
g/kg)/min,
it dilates renal and mesenteric blood vessels through stimulation of specific
dopaminergic receptors, thereby augmenting renal and mesenteric blood flow and
sodium excretion. In the range of 2 to 10 (![]() g/kg)/min,
dopamine stimulates myocardial
g/kg)/min,
dopamine stimulates myocardial ![]() 1
receptors but induces relatively little tachycardia, while at higher doses it
also stimulates
1
receptors but induces relatively little tachycardia, while at higher doses it
also stimulates ![]() -adrenergic
receptors and elevates arterial pressure.
-adrenergic
receptors and elevates arterial pressure.
Dobutamine is a synthetic catecholamine that acts on ![]() 1,
1,
![]() 2,
and
2,
and ![]() receptors.
It exerts a potent inotropic action, has only a modest cardioaccelerating
effect, and lowers peripheral vascular resistance, but since it simultaneously
raises cardiac output, it may not lower systemic arterial pressure in patients
with severe HF. Dobutamine, given in continuous infusions of 2.5 to 10 (
receptors.
It exerts a potent inotropic action, has only a modest cardioaccelerating
effect, and lowers peripheral vascular resistance, but since it simultaneously
raises cardiac output, it may not lower systemic arterial pressure in patients
with severe HF. Dobutamine, given in continuous infusions of 2.5 to 10 (![]() g/kg)/min,
is useful in the treatment of acute HF without hypotension.
g/kg)/min,
is useful in the treatment of acute HF without hypotension.
A
major problem with sympathomimetics is the loss of responsiveness, apparently
due to "downregulation" of adrenergic receptors, which becomes
evident within 8 h of continuous administration. This problem may be managed by
intermittent therapy.
Phosphodiesterase Inhibitors
These
bipyridines, amrinone and milrinone, are noncatecholamine, nonglycoside agents
that exert both positive inotropic and vasodilator actions by inhibiting a
specific phosphodiesterase. They are suitable for intravenous use only; by
simultaneously stimulating cardiac contractility and dilating the systemic
vascular bed they reverse the major hemodynamic abnormalities associated with
intractable HF. Amrinone and milrinone may be administered for the same
conditions in which sympathomimetics are useful and may be employed together
with dopamine or dobutamine.
Other Measures
Anticoagulants
Patients
with severe HF are at increased risk of pulmonary emboli secondary to venous
thrombosis and of systemic emboli secondary to intracardiac thrombi and should
be treated with warfarin. Patients with HF and atrial fibrillation, previous
venous thrombosis, and pulmonary or systemic emboli are at especially high risk
and should receive heparin followed by warfarin.
Diastolic Heart Failure
The
major goal in the treatment of this condition is to eliminate or reduce the
causes of diastolic dysfunction, such as ventricular hypertrophy, fibrosis, or
ischemia. The second is to reduce pulmonary and/or systemic venous congestion,
a major consequence of diastolic dysfunction.
Management of Arrhythmias
Premature
ventricular contractions and episodes of asymptomatic ventricular tachycardia
are common in advanced HF. Sudden death, presumably due to ventricular
fibrillation, is responsible for about one-half of all deaths in this
condition. (The remainder are due to failure of the cardiac pump.) The
management of arrhythmias should commence with correction of electrolyte and
acid-base disturbances (Chaps. 49 and 50), especially diuretic-induced
hypokalemia, as well as digitalis intoxication (see above). Treatment with
class I antiarrhythmics such as quinidine, procainamide, or flecainide (Chap.
230) is fraught with danger because these drugs are proarrhythmic in patients
with HF. Amiodarone, a class III antiarrhythmic, on the other hand, is well
tolerated and is the drug of choice for patients with heart failure and atrial
fibrillation. Patients who have been resuscitated from sudden death, those with
syncope or presyncope due to ventricular arrhythmias, and those with
asymptomatic ventricular tachyarrhythmias in whom ventricular tachycardia can
be induced during electrophysiologic testing should be considered for the
implantable automatic defibrillator. This may prevent recurrence of the
arrhythmia and sudden death; back-up pacing may prevent sudden death due to
bradyarrhythmias.
Refractory Heart Failure
When
the response to ordinary treatment is inadequate, HF is considered to be
refractory. Before assuming that this condition simply reflects advanced,
terminal, myocardial depression, careful consideration must be given to several
possibilities: (1) an underlying and overlooked cause of the heart disease that
may be amenable to specific surgical or medical therapy, such as infective
endocarditis, hypertension, thyrotoxicosis, or silent aortic or mitral stenosis;
(2) one or a combination of the precipitating causes of HF, such as pulmonary
or urinary tract infection, recurrent pulmonary emboli, arterial hypoxemia,
anemia, or arrhythmia; and (3) complications of overly vigorous therapy, such
as digitalis intoxication, hypovolemia, or electrolyte imbalance. Recognition
and proper treatment of the aforementioned complications are likely to restore
responsiveness to therapy.
Hyponatremia
is a manifestation of advanced refractory HF. It may be a complication of
overaggressive diuresis leading to reduced glomerular filtration rate and
decreased delivery of NaCl to the diluting sites in the distal tubule.
Hyponatremia may also result from nonosmotic stimuli for the continued
secretion of antidiuretic hormone. Therapy involves improvement of the
cardiovascular status, if possible (sometimes requiring the administration of a
sympathomimetic amine), as well as temporary cessation of diuretic therapy and
restriction of oral water intake. Hypertonic saline is very rarely indicated
because total-body sodium is usually elevated, not depressed, in HF.
The
combination of the intravenously administered vasodilator sodium nitroprusside,
a phosphodiesterase inhibitor (amrinone or milrinone), together with a
sympathomimetic amine (dopamine or dobutamine) often results in additive
effects, raising cardiac output and lowering filling pressure.
In
hospitalized patients with refractory HF, therapy guided by hemodynamic
measurements provided by a balloon flotation (Swan-Ganz) catheter may be helpful.
The goal of manipulating diuretics, vasodilators, and inotropic agents is to
achieve a pulmonary capillary wedge pressure of 15 to 18 mmHg, a right atrial
pressure of 5 to 8 mmHg, a cardiac index > 2.2 (L/min)/m2, and a
systemic vascular resistance of 800 to 1200 dyne ⋅ s/cm5. Once
these values are achieved, an attempt should be made to convert the patient
from intravenous to oral vasodilator therapy.
Assisted Circulation/Cardiac Transplantation
When
patients with HF become unresponsive to a combination of all the aforementioned
therapeutic measures, are in New York Heart Association class IV, and are
deemed unlikely to survive 1 year, they should be considered for temporary
assisted circulation and/or cardiac transplantation (see Chap. 233).
Treatment of Acute Pulmonary Edema
Pulmonary
edema secondary to left ventricular failure or mitral stenosis is described in
Chap. 32. It is life-threatening and must be considered a medical emergency. As
is the case for the more chronic forms of HF, in the treatment of pulmonary
edema, attention must be directed to identifying and removing any precipitating
causes of decompensation, such as an arrhythmia or infection. However, because
of the acute nature of the problem, a number of additional nonspecific measures
are necessary. If it does not delay treatment unduly, recording pulmonary
vascular pressures through a Swan-Ganz catheter and intraarterial pressure
directly is advisable. The first six measures listed below are ordinarily
applied simultaneously or nearly so.
1. Morphine is administered
intravenously repetitively, as needed, in doses from 2 to 5 mg. This drug
reduces anxiety, reduces adrenergic vasoconstrictor stimuli to the arteriolar
and venous beds, and thereby helps to break a vicious cycle. Naloxone should be
available in case respiratory depression occurs.
2. Because the alveolar
edema interferes with O2 diffusion resulting in arterial hypoxemia,
100% O2 should be administered, preferably under positive pressure.
The latter increases intraalveolar pressure, reduces transudation of fluid from
the alveolar capillaries, and impedes venous return to the thorax, reducing
pulmonary capillary pressure.
3. The patient should be
maintained in the sitting position, with the legs dangling along the side of
the bed, if possible, which tends to reduce venous return.
4. Intravenous loop
diuretics, such as furosemide or ethacrynic acid (40 to 100 mg) or bumetanide
(1 mg), will, by rapidly establishing a diuresis, reduce circulating blood
volume and thereby hasten the relief of pulmonary edema. In addition, when
given intravenously, furosemide also exerts a venodilator action, reduces
venous return, and thereby improves pulmonary edema even before the diuresis
commences.
5. Afterload reduction is
achieved with intravenous sodium nitroprusside at 20 to 30 ![]() g/min
in patients whose systolic arterial pressures exceed 100 mmHg.
g/min
in patients whose systolic arterial pressures exceed 100 mmHg.
6. Inotropic support should
be provided by dopamine or dobutamine as described on p. 1327. Patients with
systolic HF who are not receiving digitalis should receive 0.75 to 1.0 mg
digoxin intravenously over 15 min.
7. Sometimes, aminophylline
(theophylline ethylenediamine), 240 to 480 mg intravenously, is effective in
diminishing bronchoconstriction, increasing renal blood flow and sodium
excretion, and augmenting myocardial contractility.
8. If the above-mentioned
measures are not sufficient, rotating tourniquets should be applied to the
extremities.
After
these emergency measures have been instituted and the precipitating factors
treated, the diagnosis of the underlying cardiac disorder responsible for the
pulmonary edema must be established, if it is not already known. After
stabilization of the patient's condition, a long-range strategy for prevention
of future episodes of pulmonary edema must be established, and this may require
surgical treatment.
Prognosis
The
prognosis in patients with HF depends primarily on the nature of the underlying
heart disease and on the presence or absence of a precipitating factor that can
be treated. When one of the latter can be identified and removed, the outlook
for immediate survival is far better than if HF occurs without any obvious
precipitating cause. In the latter situation, survival usually ranges between 6
months and 4 years depending on the severity (Fig. 232-2). The long-term
prognosis is more favorable when the underlying forms of heart disease, e.g.,
valvular heart disease, can be treated effectively. The prognosis can be
estimated by observing the response to treatment. When clinical improvement
occurs with only modest dietary sodium restriction and small doses of
diuretics, the outlook is far better than if, in addition to these measures,
intensive diuretic therapy and vasodilators are necessary. Other factors that
have been shown to be associated with a poor prognosis include a severely
depressed ejection fraction (<25%), a reduced maximal O2 uptake
[<10 (mL/kg)/min], the inability to walk on the level and at a normal pace
for more than 3 min, reduced (<133 mEq/L) serum sodium concentration,
reduced (<3 mEq/L) serum potassium concentration, elevated circulating
atrial and brain natriuretic peptide and norepinephrine concentrations, as well
as frequent ventricular extrasystoles. A large fraction of patients with HF die
suddenly, presumably of ventricular fibrillation. Unfortunately, there is no
evidence that this complication can be prevented by the administration of
antiarrhythmic agents. See guideline material, Table 232-4.
Figure 232-2: The natural history of congestive heart
failure (CHF). Once left ventricular systolic dysfunction is present, it
usually progresses, albeit not predictably. As left ventricular dysfunction
progresses and symptoms increase, mortality rate increases and the process
becomes inexorable. Myocyte loss and fibrosis become irreversible. An effective
preventive measure must be introduced before onset or early in the course of
progressive left ventricular dysfunction.