Disorders of Ventilation
Definition and Etiology
Alveolar hypoventilation exists by definition when arterial PCO2 (PaCO2) increases above the normal range of 37 to 43 mmHg, but in clinically important hypoventilation syndromes PaCO2 is generally in the range of 50 to 80 mmHg. Hypoventilation disorders can be acute or chronic. The acute disorders, which represent life-threatening emergencies, are discussed in Chap. 265; this chapter deals with chronic hypoventilation syndromes.
Chronic hypoventilation can result from numerous disease entities (Table 263-1), but in all cases the underlying mechanism involves a defect in either the metabolic respiratory control system, the respiratory neuromuscular system, or the ventilatory apparatus. Disorders associated with impaired respiratory drive, defects in the respiratory neuromuscular system, some chest wall disorders such as obesity, and upper airway obstruction produce an increase in PaCO2, despite normal lungs, because of a reduction in overall minute volume of ventilation and hence in alveolar ventilation. In contrast, most disorders of the chest wall and disorders of the lower airways and lungs may produce an increase in PaCO2, despite a normal or even increased minute volume of ventilation, because of severe ventilation-perfusion mismatching that results in net alveolar hypoventilation.
Table 263-1: Chronic Hypoventilation Syndromes
|
Mechanism |
Site of Defect |
Disorder |
||||||||
|
Impaired respiratory drive |
Peripheral and central chemoreceptors |
Carotid body dysfunction, trauma |
||||||||
|
|
Brainstem respiratory neurons |
Bulbar poliomyelitis, encephalitis |
||||||||
|
Defective respiratory neuromuscular system |
Spinal cord and peripheral nerves |
High cervical trauma |
||||||||
|
|
Respiratory muscles |
Myasthenia gravis |
||||||||
|
Impaired ventilatory apparatus |
Chest wall |
Kyphoscoliosis |
||||||||
|
|
Airways and lungs |
Laryngeal and tracheal stenosis |
||||||||
Several hypoventilation syndromes involve combined disturbances in two elements of the respiratory system. For example, patients with chronic obstructive pulmonary disease may hypoventilate not simply because of impaired ventilatory mechanics but also because of a reduced central respiratory drive, which can be inherent or secondary to a coexisting metabolic alkalosis (related to diuretic and steroid therapy).
Physiologic and Clinical Features
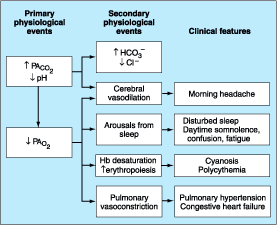
Regardless of cause, the hallmark of all alveolar hypoventilation syndromes is an increase in alveolar PCO2 (PACO2) and therefore in PaCO2 (Fig. 263-1). The resulting respiratory acidosis eventually leads to a compensatory increase in plasma HCO3- concentration and a decrease in Cl- concentration. The increase in PACO2 produces an obligatory decrease in PAO2, resulting in hypoxemia. If severe, the hypoxemia manifests clinically as cyanosis and can stimulate erythropoiesis and induce secondary polycythemia. The combination of chronic hypoxemia and hypercapnia may also induce pulmonary vasoconstriction, leading eventually to pulmonary hypertension, right ventricular hypertrophy, and congestive heart failure. The disturbances in arterial blood gases are typically magnified during sleep because of a further reduction in central respiratory drive. The resulting increased nocturnal hypercapnia may cause cerebral vasodilation leading to morning headache; sleep quality may also be severely impaired, resulting in morning fatigue, daytime somnolence, mental confusion, and intellectual impairment. Other clinical features associated with hypoventilation syndromes are related to the specific underlying disease (Table 263-1).
Figure 263-1: Physiologic and clinical features of alveolar hypoventilation. Hb, hemoglobin; PACO2, alveolar PCO2; PAO2, alveolar PO2.
Diagnosis
Investigation of the patient with chronic hypoventilation involves several laboratory tests that will usually localize the disorder to either the metabolic respiratory control system, the neuromuscular system, or the ventilatory apparatus (Fig. 263-2). Defects in the control system impair responses to chemical stimuli, including ventilatory, occlusion pressure, and diaphragmatic electromyographic (EMG) responses. During sleep, hypoventilation is usually more marked, and central apneas and hypopneas are common. However, because the behavioral respiratory control system (which is anatomically distinct from the metabolic control system), the neuromuscular system, and the ventilatory apparatus are intact, such patients can usually hyperventilate voluntarily, generate normal inspiratory and expiratory muscle pressures (PImax, PEmax, respectively) against an occluded airway, generate normal lung volumes and flow rates on routine spirometry, and have normal respiratory system resistance and compliance and a normal alveolar-arterial PO2[(A - a)PO2] difference. Patients with defects in the respiratory neuromuscular system also have impaired responses to chemical stimuli but in addition are unable to hyperventilate voluntarily or to generate normal static respiratory muscle pressures, lung volumes, and flow rates. However, at least in the early stages of the disease, the resistance and compliance of the respiratory system and the alveolar-arterial oxygen difference are normal.
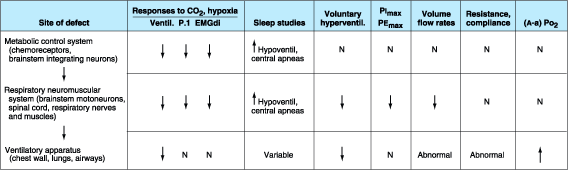
Figure 263-2: Pattern of laboratory test results in alveolar hypoventilation syndromes, based on the site of defect. Ventil, ventilation; P.1, mouth pressure generated after 0.1 s of inspiration against an occluded airway; EMGdi, diaphragmatic EMG; PImax, PEmax, maximum inspiratory or expiratory pressure that can be generated against an occluded airway; (A - a) PO2, alveolar-arterial PO2 difference; N, normal. Defects in the metabolic control system impair central respiratory drive in response to chemical stimuli (CO2 or hypoxia); therefore responses of EMGdi, P.1, and minute volume of ventilation are reduced and hypoventilation during sleep is aggravated. In contrast, tests of voluntary respiratory control, muscle strength, lung mechanics, and gas exchange [(A - a)PO2] are normal. Defects in the respiratory neuromuscular system impair muscle strength; therefore all tests dependent on muscular activity (voluntary or in response to metabolic stimuli) are abnormal, but lung resistance, lung compliance, and gas exchange are normal. Defects in the ventilatory apparatus usually impair gas exchange. Because resistance and compliance are also impaired, all tests dependent on ventilation (whether voluntary or in response to chemical stimuli) are abnormal; in contrast, tests of muscle activity or strength that do not involve airflow (i.e., P.1, EMGdi, PImax, PEmax) are normal.
In contrast to patients with disorders of the respiratory control or neuromuscular systems, patients with disorders of the chest wall, lungs, and airways typically demonstrate abnormalities of respiratory system resistance and compliance and have a widened (A - a)PO2. Because of the impaired mechanics of breathing, routine spirometric tests are abnormal, as is the ventilatory response to chemical stimuli. However, because the neuromuscular system is intact, tests that are independent of resistance and compliance are usually normal, including tests of respiratory muscle strength and of respiratory control that do not involve airflow.
Treatment
The management of chronic hypoventilation must be individualized to the patient's particular disorder, circumstances, and needs and should include measures directed toward the underlying disease. Coexistent metabolic alkalosis should be corrected, including elevations of HCO3- that are inappropriately high for the degree of chronic hypercapnia. Administration of supplemental oxygen is effective in attenuating hypoxemia, polycythemia, and pulmonary hypertension, but can aggravate CO2 retention and the associated neurologic symptoms. For this reason, supplemental oxygen must be prescribed judiciously and the results monitored carefully. Pharmacologic agents that stimulate respiration (particularly progesterone) are of benefit in some patients, but generally, results are disappointing.
Most patients with chronic hypoventilation related to impairment of respiratory drive or neuromuscular disease eventually require mechanical ventilatory assistance for effective management. When hypoventilation is severe, treatment may be required on a 24-h basis, but in most patients ventilatory assistance only during sleep produces dramatic clinical improvement and lowering of daytime PaCO2. In patients with reduced respiratory drive but intact respiratory lower motor neurons, phrenic nerves, and respiratory muscles, diaphragmatic pacing through an implanted phrenic electrode can be very effective. However, for patients with defects in the respiratory nerves and muscles, electrophrenic pacing is contraindicated. Such patients can usually be managed effectively with either intermittent negative-pressure ventilation in a cuirass or intermittent positive-pressure ventilation delivered through a tracheostomy or nose mask. For patients who require ventilatory assistance only during sleep, positive-pressure ventilation through a nose mask is the preferred method because it obviates a tracheostomy and avoids the problem of upper airway occlusion that can arise in a negative-pressure ventilator. Hypoventilation related to restrictive disorders of the chest wall (Table 263-1) can also be managed effectively with nocturnal intermittent positive-pressure ventilation through a nose mask or tracheostomy.
Hypoventilation Syndromes
Primary Alveolar Hypoventilation
Primary alveolar hypoventilation (PAH) is a disorder of unknown cause characterized by chronic hypercapnia and hypoxemia in the absence of identifiable neuromuscular disease or mechanical ventilatory impairment. The disorder is thought to arise from a defect in the metabolic respiratory control system, but few neuropathologic studies have been reported in such patients. Recent studies in animals suggest an important role for genetic factors in the pathogenesis of hypoventilation. Isolated PAH is relatively rare, and although it occurs in all age groups, the majority of reported cases have been in males aged 20 to 50 years. The disorder typically develops insidiously and often first comes to attention when severe respiratory depression follows administration of standard doses of sedatives or anesthetics. As the degree of hypoventilation increases, patients typically develop lethargy, fatigue, daytime somnolence, disturbed sleep, and morning headaches; eventually cyanosis, polycythemia, pulmonary hypertension, and congestive heart failure occur (Fig. 263-1). Despite severe arterial blood gas derangements, dyspnea is uncommon, presumably because of impaired chemoreception and ventilatory drive. If left untreated, PAH is usually progressive over a period of months to years and ultimately fatal.
The key diagnostic finding in PAH is a chronic respiratory acidosis in the absence of respiratory muscle weakness or impaired ventilatory mechanics (Fig. 263-2). Because patients can hyperventilate voluntarily and reduce PaCO2 to normal or even hypocapnic levels, hypercapnia may not be demonstrable in a single arterial blood sample, but the presence of an elevated plasma HCO3- level should draw attention to the underlying chronic disturbance. Despite normal ventilatory mechanics and respiratory muscle strength, ventilatory responses to chemical stimuli are reduced or absent (Fig. 263-2), and breath-holding time may be markedly prolonged without any sensation of dyspnea.
Patients with PAH maintain rhythmic respiration when awake, although the level of ventilation is below normal. However, during sleep, when breathing is critically dependent on the metabolic control system, there is typically a further deterioration in ventilation with frequent episodes of central hypopnea or apnea.
PAH must be distinguished from other central hypoventilationsyndromes that are secondary to underlying neurologic disease of the brainstem or chemoreceptors (Table 263-1). This distinction requires a careful neurologic investigation for evidence of brainstem or autonomic disturbances. Unrecognized respiratory neuromuscular disorders, particularly those that produce diaphragmatic weakness, are often misdiagnosed as PAH. However, such disorders can usually be suspected on clinical grounds (see below) and can be confirmed by the finding of reduced voluntary hyperventilation, as well as PImax and PEmax.
Some patients with PAH respond favorably to respiratory stimulant medications and to supplemental oxygen. However, the majority eventually require mechanical ventilatory assistance. Excellent long-term benefits can be achieved with diaphragmatic pacing by electrophrenic stimulation or with negative- or positive-pressure mechanical ventilation. The administration of such treatment only during sleep is sufficient in most patients.
Respiratory Neuromuscular Disorders
Several primary disorders of the spinal cord, peripheral respiratory nerves, and respiratory muscles produce a chronic hypoventilation syndrome (Table 263-1). Hypoventilation usually develops gradually over a period of months to years and often first comes to attention when a relatively trivial increase in mechanical ventilatory load (such as mild airways obstruction) produces severe respiratory failure. In some of the disorders (such as motor neuron disease, myasthenia gravis, and muscular dystrophy), involvement of the respiratory nerves or muscles is usually a later feature of a more widespread disease. In other disorders, respiratory involvement can be an early or even isolated feature, and hence the underlying problem is often not suspected. Included in this category are the postpolio syndrome (a form of chronic respiratory insufficiency that develops 20 to 30 years following recovery from poliomyelitis), the myopathy associated with adult acid maltase deficiency, and idiopathic diaphragmatic paralysis.
Generally, respiratory neuromuscular disorders do not result in chronic hypoventilation unless there is significant weakness of the diaphragm. Distinguishing features of bilateral diaphragmatic weakness include orthopnea, paradoxical movement of the abdomen in the supine posture, and paradoxical diaphragmatic movement under fluoroscopy. However, the absence of these features does not exclude diaphragmatic weakness. Important laboratory features are a rapid deterioration of ventilation during a maximum voluntary ventilation maneuver and reduced PImax and PEmax (Fig. 263-2). More sophisticated investigations reveal reduced or absent transdiaphragmatic pressures, calculated from simultaneous measurement of esophageal and gastric pressures; reduced diaphragmatic EMG responses (recorded from an esophageal electrode) to transcutaneous phrenic nerve stimulation; and marked hypopnea and arterial oxygen desaturation during rapid eye movement sleep, when there is normally a physiologic inhibition of all nondiaphragmatic respiratory muscles and breathing becomes critically dependent on diaphragmatic activity.
The management of chronic alveolar hypoventilation due to respiratory neuromuscular disease involves treatment of the underlying disorder, where feasible, and mechanical ventilatory assistance as described for the primary alveolar hypoventilation syndrome. However, electrophrenic diaphragmatic pacing is contraindicated in these disorders, except for high cervical spinal cord lesions in which the phrenic lower motor neurons and nerves are intact.
Obesity-Hypoventilation Syndrome
Massive obesity represents a mechanical load to the respiratory system because the added weight on the rib cage and abdomen serves to reduce the compliance of the chest wall. As a result, the functional residual capacity (i.e., end-expiratory lung volume) is reduced, particularly in the recumbent posture. An important consequence of breathing at a low lung volume is that some airways, particularly those in the lung bases, may be closed throughout part or even all of each tidal breath, resulting in underventilation of the lung bases and widening of the (A - a)PO2. Nevertheless, in the majority of obese individuals, central respiratory drive is increased sufficiently to maintain a normal PaCO2. However, a small proportion of obese patients develop chronic hypercapnia, hypoxemia, and eventually polycythemia, pulmonary hypertension, and right-sided heart failure. Recent studies in mice demonstrate that genetically obese mice lacking circulating leptin also develop chronic hypoventilation that can be reversed by leptin infusions. Those patients who also develop daytime somnolence have been designated as having the Pickwickian syndrome (Chap. 27). In many such patients, obstructive sleep apnea is a prominent feature, and even in those patients without sleep apnea, sleep-induced hypoventilation is an important element of the disorder and contributes to its progression. Most patients demonstrate a decrease in central respiratory drive, which may be inherent or acquired, and many have mild to moderate degrees of airflow obstruction, usually related to smoking. Based on these considerations, several therapeutic measures can be of considerable benefit, including weight loss, cessation of smoking, elimination of obstructive sleep apnea, and enhancement of respiratory drive by medications such as progesterone.
Hyperventilation and Its Syndromes
Definition and Etiology
Alveolar hyperventilation exists when PaCO2 decreases below the normal range of 37 to 43 mmHg. Hyperventilation is not synonymous with hyperpnea, which refers to an increased minute volume of ventilation without reference to PaCO2. Although hyperventilation is frequently associated with dyspnea, patients who are hyperventilating do not necessarily complain of shortness of breath; and conversely, patients with dyspnea need not be hyperventilating.
Numerous disease entities can be associated with alveolar hyperventilation (Table 263-2), but in all cases the underlying mechanism involves an increase in respiratory drive that is mediated through either the behavioral or the metabolic respiratory control systems (Fig. 263-3). Thus hypoxemia drives ventilation by stimulating the peripheral chemoreceptors, and several pulmonary disorders and congestive heart failure drive ventilation by stimulating afferent vagal receptors in the lungs and airways. Low cardiac output and hypotension stimulate the peripheral chemoreceptors and inhibit the baroreceptors, both of which increase ventilation. Metabolic acidosis, a potent respiratory stimulant, excites both the peripheral and central chemoreceptors and increases the sensitivity of the peripheral chemoreceptors to coexistent hypoxemia. Hepatic failure can also produce hyperventilation, presumably as a result of metabolic stimuli acting on the peripheral and central chemoreceptors.
Figure 263-3: Schematic diagram of the mechanisms involved in alveolar hyperventilation.
Table 263-2: Hyperventilation Syndromes
Hypoxemia
Pulmonary disorders
Cardiovascular disorders
Metabolic disorders
Neurologic and psychogenic disorders
Drug-induced
Miscellaneous
Several neurologic and psychological disorders are thought to drive ventilation through the behavioral respiratory control system. Included in this category are psychogenic or anxiety hyperventilation and severe cerebrovascular insufficiency, which may interfere with the inhibitory influence normally exerted by cortical structures on the brainstem respiratory neurons. Rarely, disorders of the midbrain and hypothalamus induce hyperventilation, and it is conceivable that fever and sepsis also cause hyperventilation through effects on these structures. Several drugs cause hyperventilation by stimulating the central or peripheral chemoreceptors or by direct action on the brainstem respiratory neurons. Chronic hyperventilation is a normal feature of pregnancy and results from the effects of progesterone and other hormones acting on the respiratory neurons.
Physiologic and Clinical Features
Because hyperventilation is associated with increased respiratory drive, muscle effort, and minute volume of ventilation, the most frequent symptom associated with hyperventilation is dyspnea. However, there is considerable discrepancy between the degree of hyperventilation, as measured by PaCO2, and the degree of associated dyspnea. From a physiologic standpoint, hyperventilation is beneficial in patients who are hypoxemic, because the alveolar hypocapnia is associated with an increase in alveolar and arterial PO2. Conversely, hyperventilation can also be detrimental. In particular, the alkalemia associated with hypocapnia may produce neurologic symptoms, including dizziness, visual impairment, syncope, and seizure activity (secondary to cerebral vasoconstriction); parasthesia, carpopedal spasm, and tetany (secondary to decreased free serum calcium); and muscle weakness (secondary to hypophosphatemia). Severe alkalemia can also induce cardiac arrhythmias and evidence of myocardial ischemia. Patients with a primary respiratory alkalosis are also prone to periodic breathing and central sleep apnea (Chap. 264).
Diagnosis
In most patients with a hyperventilation syndrome, the cause is readily apparent on the basis of history, physical examination, and knowledge of coexisting medical disorders (Table 263-2). In patients in whom the cause is not clinically apparent, investigation begins with arterial blood gas analysis, which establishes the presence of alveolar hyperventilation (decreased PaCO2) and its severity. Equally important is the arterial pH, which generally allows the disorder to be classified as either a primary respiratory alkalosis (elevated pH) or a primary metabolic acidosis (decreased pH). Also of importance is the PaO2 and calculation of the (A - a)PO2, since a widened alveolar-arterial oxygen difference suggests a pulmonary disorder as the underlying cause. The finding of a reduced plasma HCO3- level establishes the chronic nature of the disorder and points toward an organic cause. Measurements of ventilation and arterial or transcutaneous PCO2 during sleep are very useful in suspected psychogenic hyperventilation, since such patients do not maintain the hyperventilation during sleep.
The disorders that most frequently give rise to unexplained hyperventilation are pulmonary vascular disease (particularly chronic or recurrent thromboembolism) and psychogenic or anxiety hyperventilation. Hyperventilation due to pulmonary vascular disease is associated with exertional dyspnea, a widened (A - a)PO2 and maintenance of hyperventilation during exercise. In contrast, patients with psychogenic hyperventilation typically complain of dyspnea at rest and not during mild exercise and of the need to sigh frequently. They are also more likely to complain of dizziness, sweating, palpitations, and paresthesia. During mild to moderate exercise, their hyperventilation tends to disappear and (A - a)PO2 is normal, but heart rate and cardiac output may be increased relative to metabolic rate.
Treatment
Alveolar hyperventilation is usually of relatively minor clinical consequence and therefore is generally managed by appropriate treatment of the underlying cause. In the few patients in whom alkalemia is thought to be inducing significant cerebral vasoconstriction, paresthesia, tetany, or cardiac disturbances, inhalation of a low concentration of CO2 can be very beneficial. For patients with disabling psychogenic hyperventilation, careful explanation of the basis of their symptoms can be reassuring and is often sufficient. Others have benefited from b -adrenergic antagonists or an exercise program. Specific treatment for anxiety may also be indicated.
Sleep Apnea
Definition and Classification
Sleep apnea is defined as an intermittent cessation of airflow at the nose and mouth during sleep. By convention, apneas of at least 10 s duration have been considered important, but in most patients the apneas are 20 to 30 s in duration and may be as long as 2 to 3 min. Sleep apnea syndrome refers to a clinical disorder that arises from recurrent apneas during sleep. The clinical importance of sleep apnea arises from the fact that it is one of the leading causes of excessive daytime sleepiness. Indeed, epidemiologic studies have established a prevalence of clinically important sleep apnea of at least 2% in middle-aged women and 4% in middle-aged men.
Sleep apneas can be central or obstructive in type. In central sleep apnea (CSA) the neural drive to all the respiratory muscles is transiently abolished. In contrast, in obstructive sleep apnea (OSA) airflow ceases despite continuing respiratory drive because of occlusion of the oropharyngeal airway.
Obstructive Sleep Apnea
Pathogenesis
The definitive event in OSA is occlusion of the upper airway usually at the level of the oropharynx. The resulting apnea leads to progressive asphyxia until there is a brief arousal from sleep, whereupon airway patency is restored and airflow resumes. The patient then returns to sleep, and the sequence of events is repeated, often up to 400 to 500 times per night, resulting in marked fragmentation of sleep.
The immediate factor leading to collapse of the upper airway in OSA is the generation of a critical subatmospheric pressure during inspiration that exceeds the ability of the airway dilator and abductor muscles to maintain airway stability. During wakefulness, upper airway muscle activity is greater than normal in patients with OSA, presumably to compensate for airway narrowing (see below) and a high upper airway resistance. Sleep plays a permissive but crucial role by reducing the activity of the muscles and their protective reflex response to subatmospheric airway pressures. Alcohol is frequently an important cofactor because of its selective depressant influence on the upper airway muscles and on the arousal response that terminates each apnea. In most patients the patency of the airway is also compromised structurally and therefore predisposed to occlusion. In a minority of patients the structural compromise is due to obvious anatomic disturbances, such as adenotonsillar hypertrophy, retrognathia, and macroglossia. However, in the majority of patients the structural defect is simply a subtle reduction in airway size that can often be appreciated clinically as "pharyngeal crowding" and that can usually be demonstrated by imaging and acoustic reflection techniques. Obesity frequently contributes to the reduction in size of the upper airways, either by increasing fat deposition in the soft tissues of the pharynx or by compressing the pharynx by superficial fat masses in the neck. More sophisticated studies also demonstrate a high airway compliance-i.e., the airway is "floppy" and therefore prone to collapse.
Pathophysiologic and Clinical Features
The narrowing of the upper airways during sleep, which predisposes to OSA, inevitably results in snoring. In most patients, snoring antedates the development of obstructive events by many years. However, the majority of snoring individuals do not have an OSA disorder, nor is there definitive evidence that snoring per se is associated with long-term health risks. Hence, in the absence of other symptoms, snoring alone does not warrant an investigation for OSA but does call for preventive counselling, particularly with regard to weight gain and alcohol consumption.
The recurrent episodes of nocturnal asphyxia and of arousal from sleep that characterize OSA lead to a series of secondary physiologic events, which in turn give rise in some patients to the clinical complications of the syndrome (Fig. 264-1). The most common manifestations are neuropsychiatric and behavioral disturbances that are thought to arise from the fragmentation of sleep and loss of slow-wave sleep induced by the recurrent arousal responses. Nocturnal cerebral hypoxia may also play an important role. The most pervasive manifestation is excessive daytime sleepiness. Initially, daytime sleepiness manifests under passive conditions, such as reading or watching television; but as the disorder progresses, sleepiness encroaches into all daily activities and can become disabling and dangerous. Several studies have demonstrated two to seven times more motor vehicle accidents in patients with OSA compared with other drivers. Other related symptoms include intellectual impairment, memory loss, and personality disturbances.
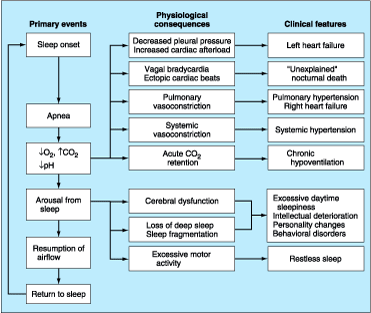 Figure 264-1: The primary sequence of events, physiologic responses, and clinical features of obstructive sleep apnea.
Figure 264-1: The primary sequence of events, physiologic responses, and clinical features of obstructive sleep apnea.
The other major manifestations of OSA are cardiorespiratory in nature and are thought to arise from the recurrent episodes of nocturnal asphyxia and of negative intrathoracic pressure, which increases left ventricular afterload (Fig. 264-1). Many patients demonstrate a cyclical slowing of the heart during the apneas to 30 to 50 beats per minute, followed by a tachycardia of 90 to 120 beats per minute during the ventilatory phase. A small number of patients develop severe bradycardia or dangerous tachyarrhythmias, leading to the notion that OSA may result in sudden death during sleep, but firm corroborative data are lacking. Unlike in healthy subjects, in patients with OSA systemic blood pressure fails to decrease during sleep. In fact, blood pressure typically rises abruptly at the termination of each obstructive event as a result of sympathetic nervous activation and reflex vasoconstriction. Furthermore, over 50% of patients with OSA have systemic hypertension. Several epidemiologic studies have implicated OSA as a risk factor for the development of systemic hypertension, and recent studies in an animal model demonstrate directly that OSA can cause sustained increases in daytime blood pressure. Emerging data also suggest that OSA can precipitate myocardial ischemia in patients with coronary artery disease and can adversely affect left ventricular function, both acutely and chronically, in patients with congestive heart failure. This complication is probably due to the combined effects of increased left ventricular afterload during each obstructive event, secondary to increased negative intrathoracic pressure (Fig. 264-1), recurrent nocturnal hypoxemia, and chronically elevated sympathoadrenal activity. Treatment of OSA in such patients often results in dramatic improvement in left ventricular function and in clinical cardiac status. Finally, up to 20% of patients with OSA develop mild pulmonary hypertension (in the absence of intrinsic lung disease), and a small proportion (<10%) develop pulmonary hypertension, right ventricular failure, polycythemia, and chronic hypercapnia and hypoxemia. All such patients have evidence of sustained daytime hypoxemia in addition to the nocturnal ventilatory disturbance, usually as a result of reduced ventilatory drive and/or diffuse airways obstruction.
Diagnosis
Although OSA occurs at any age, and is more prevalent in women than was previously thought, the typical patient is a male aged 30 to 60 years who presents with a history of snoring, excessive daytime sleepiness, nocturnal choking or gasping, witnessed apneas during sleep, moderate obesity, and often mild to moderate hypertension. The definitive investigation for suspected OSA is polysomnography, a detailed overnight sleep study that includes recording of (1) electrographic variables (electroencephalogram, electrooculogram, and submental electromyogram) that permit the identification of sleep and its various stages, (2) ventilatory variables that permit the identification of apneas and their classification as central or obstructive, (3) arterial O2 saturation by ear or finger oximetry, and (4) heart rate. Continuous measurement of transcutaneous PCO2 (which reflects arterial PCO2) can also be very useful, particularly in patients with CSA. The key diagnostic finding in OSA is episodes of airflow cessation at the nose and mouth despite evidence of continuing respiratory effort. By the time most patients come to clinical attention they have at least 10 to 15 obstructive events per hour of sleep. However, recent data suggest that a high upper airway resistance during sleep (manifested by snoring) that is accompanied by recurrent arousals from sleep, even in the absence of apneas and hypopneas, can result in a clinically important sleep-related syndrome. Therefore, the absence of outright apneas and hypopneas in a symptomatic patient may not definitely exclude a sleep-related respiratory disorder.
Because polysomnography is a time-consuming and expensive test, there is considerable interest in the role of simplified, unattended, ambulatory sleep monitoring for the investigation of OSA that would allow the patient to be studied at home, rather than in the sleep laboratory. The most useful test in this context is the recording of arterial O2 saturation by oximetry. However, the reliability of overnight oximetry in the diagnosis of OSA is dependent on the pretest probability of the disorder. In patients with a high pretest probability (based on a history of daytime sleepiness, habitual snoring, nocturnal choking or gasping, and witnessed apneas during sleep), overnight oximetry can be used to confirm the diagnosis by demonstrating recurrent episodes of arterial O2 desaturation (at a rate of at least 10 to 15 events per hour). Such findings obviate the need for full polysomnography and allow initiation of treatment with nasal continuous positive airway pressure (CPAP) during sleep (see "Treatment"). However, negative results in a patient with a high clinical probability of OSA do not exclude the diagnosis but mandate that the patient proceed to polysomnography to investigate the cause of the daytime sleepiness. In contrast, when the pretest probability of OSA is low (such as the patient with only occasional snoring, few witnessed apneas, and no daytime sleepiness), the absence of arterial O2 desaturation can be used to exclude the diagnosis and thereby obviate the need for full polysomnography.
Studies suggest that overnight oximetry can obviate the need for polysomnography in about one-third of clinic patients referred for consideration of OSA, either by confirming the diagnosis in patients with a high pretest probability of the disorder, or by excluding the diagnosis in patients with a low pretest probability. In the remaining two-thirds of patients with an intermediate pretest probability of OSA, overnight oximetry alone will not be definitive; hence such patients will require polysomnography
Treatment
(Table 264-1) Several approaches to treatment of OSA have been advocated, based on an understanding of the mechanisms underlying the disorder. Mild to moderate OSA can often be managed effectively by modest weight reduction, avoidance of alcohol, improvement of nasal patency, and avoidance of sleeping in the supine posture. Intraoral appliances, designed to keep the mandible and tongue forward, are also effective in 55 to 80% of patients. The most widely used treatments in severe OSA are uvulopalatopharyngoplasty and nasal CPAP during sleep. Uvulopalatopharyngoplasty is a surgical procedure designed to increase the pharyngeal lumen by resecting redundant soft tissue. When applied to unselected patients with OSA, it produces long-term cure in fewer than 50% but more discriminating selection of patients yields a higher rate of success. Other surgical approaches, including mandibular advancement and hyoid osteotomy have a more limited application but higher rate of success in selected patients. Nasal CPAP, which prevents upper airway occlusion by splinting the pharyngeal airway with a positive pressure delivered through a nose mask, is currently the most successful long-term approach to treatment, being well tolerated and effective in over 80% of patients, provided that they have received proper training. Patients who are unable to tolerate conventional nasal CPAP may respond to newer generation devices that provide more flexibility in adjusting the timing and levels of inspiratory and expiratory pressure cycles. For patients with ischemic heart disease or congestive heart failure who also have OSA, nasal CPAP is the only treatment that has been specifically tested and is considered the treatment of choice. Finally, for the few patients with severe OSA in whom all other treatment approaches have failed, tracheostomy can provide immediate relief, but in most centers is performed only very rarely.
Table 264-1: Management of Obstructive Sleep Apnea (OSA)
|
Mechanism |
Mild to Moderate OSA |
Moderate to Severe OSA |
|
Upper airway muscle tone |
Avoidance of alcohol, sedatives |
- |
|
Upper airway lumen size |
Weight reduction |
Uvulopalatopharyngoplasty |
|
¯ Upper airway subatmospheric pressure |
Improved nasal patency |
Nasal continuous positive airway pressure |
|
Bypass occlusion |
|
Tracheostomy |
Central Sleep Apnea
Pathogenesis
The definitive event in CSA is transient abolition of central drive to the ventilatory muscles. The resulting apnea leads to a primary sequence of events similar to those of OSA (Fig. 264-1). Several underlying mechanisms can result in cessation of respiratory drive during sleep (Table 264-2). First are defects in the metabolic respiratory control system and respiratory neuromuscular apparatus. Such defects usually produce a chronic alveolar hypoventilation syndrome (in addition to CSA) that becomes more severe during sleep when the stimulatory effect of wakefulness on breathing is abolished. In contrast are CSA disorders that arise from transient instabilities in an otherwise intact respiratory control system. Common to all these disorders is a PCO2 level during sleep that falls transiently below the critical PCO2 required for respiratory rhythm generation. The most frequent instability of this type occurs at sleep onset, because the PCO2 level of wakefulness is often lower than that required for rhythm generation in sleep; hence with loss of the stimulatory effect of wakefulness on breathing (referred to as the waking neural drive), an apnea develops at sleep onset until PCO2 rises to the critical level (Fig. 264-2). However, if the central nervous system state fluctuates at sleep onset between "asleep" and "awake," a pattern of periodic breathing develops as respiration follows the changes in state. During each cycle, the waning phase of ventilation includes an hypopnea or outright central apnea (Cheyne-Stokes respiration). In most patients with CSA, the tendency to develop periodic breathing and central apneas during sleep is enhanced by some degree of chronic hyperventilation during wakefulness that drives the PCO2 level below the threshold required for rhythm generation during sleep. Such hyperventilation is frequently idiopathic in nature. Hypoxia, whether due to high altitude or to underlying cardiorespiratory disease, also enhances the tendency to periodic breathing and CSA for the same reasons. Periodic breathing and CSA are also common in patients with congestive heart failure. In such patients the decreases in PaCO2 that trigger transient abolition of central respiratory drive are associated with higher left ventricular end-diastolic volume and filling pressure than in congestive heart failure patients without CSA. The hyperventilation probably results, therefore, from pulmonary congestion and stimulation of pulmonary vagal receptors.
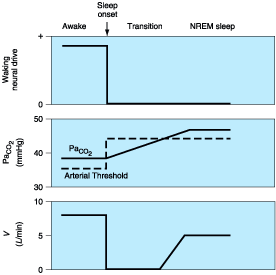
Figure 264-2: Schematic diagram of the mechanisms underlying central sleep apnea at sleep onset. With loss of the waking neural drive to breathing, the arterial threshold PCO2 for rhythm generation increases above the PaCO2 present during wakefulness; ventilation (V) falls to zero and apnea ensues until PaCO2 rises above the threshold for rhythm generation during sleep. NREM, non-rapid eye movement.
Table 264-2: Mechanisms Underlying Central Sleep Apnea
|
Underlying Mechanism |
Clinical Example |
|||||
|
Defects in metabolic control system or respiratory muscles |
Primary and secondary central alveolar hypoventilation syndromes |
|||||
|
Transient instabilities in central respiratory drive |
Sleep onset |
|||||
Pathophysiologic and Clinical Features
Many healthy individuals demonstrate a small number of central apneas during sleep, particularly at sleep onset and in rapid eye movement sleep. These apneas are not associated with any physiologic or clinical disturbances. In patients with clinically important CSA, the primary sequence of events that characterizes the disorder leads to prominent physiologic and clinical consequences (Fig. 264-1). In those patients whose CSA is a component of an alveolar hypoventilation syndrome, daytime hypercapnia and hypoxemia are usually evident, and the clinical picture is dominated by a history of recurrent respiratory failure, polycythemia, pulmonary hypertension, and right-sided heart failure. Complaints of sleeping poorly, morning headache, and daytime fatigue and sleepiness are also prominent. In contrast, in patients whose CSA results from an instability in respiratory drive, the clinical picture is dominated by features related to sleep disturbance, including recurrent nocturnal awakenings, morning fatigue, and daytime sleepiness. In patients with congestive heart failure, CSA can be an important (and frequently overlooked) cause of daytime sleepiness and fatigue. Recent studies also indicate that CSA can trigger sympathetic nervous activation in patients with heart failure and thereby exert a secondary deleterious effect on the underlying cardiac disorder.
Diagnosis
Initially, many patients with CSA are suspected clinically of having OSA because of a history of snoring, sleep disturbance, and daytime sleepiness. However, obesity and hypertension are less prominent in CSA than in OSA. Definitive diagnosis of CSA requires a polysomnographic study, with the key observation being recurrent apneas that are not accompanied by respiratory effort. Measurements of transcutaneous PCO2 are particularly useful in CSA. Those patients with a defect in respiratory control or neuromuscular function typically demonstrate an elevated PCO2 that tends to increase progressively during the night, particularly during rapid eye movement sleep. In contrast, patients with instabilities in the respiratory control system typically demonstrate a mild degree of hypocapnia, which is an integral pathogenetic feature of their disorder (see above).
Treatment
The management of patients whose CSA is a component of an alveolar hypoventilation syndrome is essentially the same as management of the underlying hypoventilation disorder (Chap. 263). Management of patients whose CSA arises from an instability of respiratory drive is more problematic. Patients with hypoxemia usually respond favorably to nocturnal supplemental oxygen. Others have responded to acidification with acetazolamide, and recent reports indicate a good response to nasal CPAP (as for OSA). The mechanism by which CPAP abolishes central apneas probably involves a small increase in PaCO2 as a result of the added expiratory mechanical load. In patients whose CSA is secondary to congestive heart failure, CPAP is particularly effective in improving sleep quality and daytime cardiac function. In fact, recent randomized trials have demonstrated that CPAP has a beneficial effect on several surrogate markers of mortality in patients with congestive heart failure, including left ventricular ejection fraction, functional mitral regurgitation, and norepinephrine concentrations.
Acute Respiratory Distress Syndrome
Lung injury in acute respiratory distress syndrome (ARDS) is characterized by increased permeability of the alveolar-capillary membrane, diffuse alveolar damage, and the accumulation of proteinaceous pulmonary edema. This clinical syndrome was first described in the archival literature by military physicians when respiratory failure occurred in battlefield casualties during World Wars I and II. However, it was not until the 1960s, when mechanical ventilation was used for patients with acute respiratory failure, that ARDS was first officially named. Initially the "A" in ARDS stood for "adult" to differentiate this syndrome from the infantile respiratory distress syndrome. With the more recent recognition that ARDS occurs in all age groups, the "A" now stands for "acute."
The diagnostic criteria used to define ARDS have evolved over the past three decades. Originally, most definitions required three general criteria: severe hypoxemia, decreased pulmonary compliance, and diffuse pulmonary infiltrates on chest radiograph. With the increasing utilization of pulmonary arterial catheters in the intensive care unit, ARDS was noted to be a "noncardiogenic" form of pulmonary edema (Chap. 32). Subsequently, some proposed definitions of ARDS required documentation of a normal pulmonary arterial occlusion pressure. However, due to the lack of an established definition and the recognition that ARDS is the severe form of a wide spectrum of lung injury, an American-European Consensus Conference proposed a new definition of ARDS that is now uniformly accepted (Table 265-1). Acute lung injury, which is a mild form of ARDS, was also defined and differs from ARDS based on less severe hypoxemia (Table 265-1).
Table 265-1: Recommended Criteria for Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS)
|
|
Timing |
Oxygenation |
Chest Radiograph |
Pulmonary Arterial Occlusion Pressure |
||||||||||||||
|
ALI Criteria |
Acute onset |
PaO2/FIO2 <300 mmHg (regardless of PEEP level) |
Bilateral infiltrates seen on frontal chest radiograph |
< 18mmHg when measured or no clinical evidence of left atrial hypertension |
||||||||||||||
|
ARDS Criteria |
Acute onset |
PaO2/FIO2 <200 mmHg (regardless of PEEP level) |
Bilateral infiltrates seen on frontal chest radiograph |
< 18 mmHg when measured or no clinical evidence of left atrial hypertension |
||||||||||||||
|
|
|
|
||||||||||||||||
Clinical Characteristics
Many predisposing factors are associated with the development of ARDS, including conditions that injure the lung directly and those that produce damage through indirect mechanisms via the hematogenous delivery of inflammatory mediators (Table 265-2). The most common of these at-risk conditions are severe sepsis, major trauma, and aspiration of gastric contents. In general, 30 to 40% of individuals with at least one of these diagnoses will eventually develop ARDS. This incidence increases in patients with more than one at-risk condition. A history of chronic alcohol abuse is also associated with an increased risk of developing ARDS in critically ill patients with an at-risk diagnosis.
Table 265-2: Conditions That May Lead to the Acute Respiratory Distress Syndrome
|
Direct injury to alveolar epithelium |
Indirect lung injury |
ARDS occurs within 5 days of the initial at-risk diagnosis in the majority of patients, and over 50% will develop ARDS in the first 24 h. The earliest clinical sign is often an increase in the respiratory frequency, followed by dyspnea. There are no characteristic laboratory abnormalities for ARDS patients except those related to a specific underlying condition, such as leukocytosis in sepsis or an elevated serum amylase level in pancreatitis. Radiographically, the lung fields may be clear initially; diffuse bilateral interstitial or alveolar infiltrates occur as ARDS develops (Fig. 265-1). Though these radiographic changes appear homogeneous on chest radiograph, computed tomography demonstrates a heterogeneous pattern with a predominance of infiltrates in the dependent regions of the lung (Fig. 265-2).
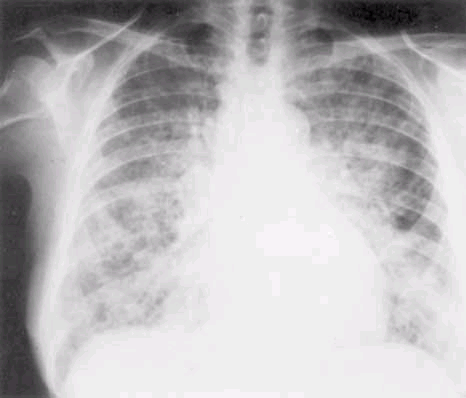
Figure 265-1: A standard posteroanterior chest radiograph from a patient with acute respiratory distress syndrome secondary to a severe viral pneumonitis. Such a diffuse radiographic change is typical of all conditions listed in Table 265-2 when they are severe enough to cause acute hypoxemic respiratory failure. A similar radiographic picture is also seen in pulmonary edema due to left ventricular failure (Chap. 32).
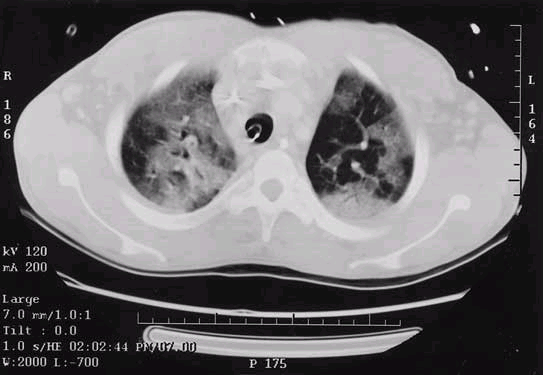
Figure 265-2: Computed tomographic image of a patient with acute respiratory distress syndrome demonstrating marked heterogeneity of pulmonary infiltrates with increased density in dependent regions.
Pathophysiology
ARDS may be the pulmonary manifestation of a systemic process and is the consequence of an overexpression of the normal inflammatory response. This inflammatory cascade has been divided into three overlapping phases-initiation, amplification, and injury. During initiation, a precipitating event, such as sepsis, causes both immune and nonimmune cells to produce and release a variety of mediators and cytokines, such as tumor necrosis factor and interleukin 1. Subsequently, during amplification, effector cells, such as neutrophils, are activated, recruited, and retained in specific target organs including the lung. Interleukin 8, which is produced by monocytes and other cell types, appears to play an important role in neutrophil activation. Once the effector cells have been sequestered in the lung, they then release reactive oxygen metabolites and proteases, causing cellular damage during the injury phase. This inflammatory cascade can occur systemically and therefore may alter the function of many organ systems-a clinical entity called multiple organ dysfunction syndrome.
The pathophysiologic hallmark of ARDS is increased vascular permeability to proteins, so that even mild elevations of pulmonary capillary pressures (due to increased intravenous liquid administration and/or myocardial depression, which may occur in sepsis) greatly increase interstitial and alveolar edema. Alveolar damage is further exaggerated by the quantitative reduction is surfactant synthesis due to injury to type II pneumocytes as well as to further qualitative abnormalities in the size, composition, and metabolism of the remaining surfactant pool, leading to alveolar collapse. Although these atelectatic and liquid-filled regions of the lung contribute to a reduction in the compliance of the lung as a whole, significant regions of nondependent lung have relatively normal mechanical and gas-exchanging properties. However, the decreased overall pulmonary compliance requires large inspiratory pressures to be generated by the respiratory muscles, resulting in an increase in the work of breathing.
Though ARDS is not routinely considered a disease of the airways, airway resistance may be increased due to bronchial wall edema and cytokine-mediated bronchospasm. Pulmonary vascular resistance and pulmonary arterial pressures may also be elevated as a result of increased pulmonary vascular smooth-muscle tone, perivascular edema, microvascular thrombosis, and the production of humoral factors such as leukotrienes and thromboxane A2, which can directly cause vasoconstriction.
Pathology
During the initial exudative phase, covering the first few days after lung injury, the following occur: (1) epithelial cell injury represented by extensive necrosis of type I pneumocytes and a denuded basement membrane, (2) swelling of endothelial cells with the widening of intercellular junctions, (3) the formation of hyaline membranes composed of fibrin and other matrix proteins in alveolar ducts and airspaces, and (4) a neutrophilic inflammation. Fibrin thrombi may be seen in the alveolar capillaries and smaller pulmonary arteries. The second pathologic phase of ARDS is characterized by proliferation of a variety of cells and resolution of the neutrophilic inflammation. Cuboidal type II cells and squamous epithelium cover denuded alveolar basement membranes. Over the ensuing days to weeks, architectural restoration of lung tissue is usually observed in survivors of ARDS. However, interstitial fibrosis and extensive restructuring of the lung parenchyma may occur with cystic and honeycomb changes in some ARDS patients, resulting in chronic pulmonary dysfunction or death.
Treatment
Currently there are no specific therapies that correct the underlying abnormalities in the permeability of the alveolar-capillary membrane or control the activated inflammatory response in patients with ARDS. However, the use of physiologically targeted strategies of mechanical ventilation and intensive care unit management have led to a more favorable outcome for these critically ill patients.
Mechanical Ventilatory Support
In the presence of ARDS, adequate oxygenation is not usually maintained when oxygen is supplied through noninvasive measures. Therefore, most ARDS patients require mechanical ventilation during their hospitalization. The primary goal of the ventilatory management in ARDS is to achieve ventilation and oxygenation that are adequate to support organ function. The major complications of mechanical ventilation are oxygen toxicity and barotrauma, which include not only pneumothorax, pneumomedistinum, and subcutaneous emphysema but also primary alveolar damage. As demonstrated on computed tomography images of the lungs in ARDS patients (Fig. 265-2), a large portion of the alveoli are atelectatic or liquid-filled. However, some nondependent regions of the lung remain radiographically unaffected, and due to their greater compliance they receive a greater proportion of the tidal volume. When large tidal volumes (10 to 12 mL/kg of ideal body weight) are forced into these smaller areas, damage may occur in epithelial and endothelial cells. The sequelae of this injury include alterations in lung liquid balance, increases in permeability, and severe alveolar damage. The deleterious effects of these large tidal volumes and subsequent high alveolar pressures has been termed volutrauma.
The currently recommended ventilatory strategies for ARDS patients focus on the limitation of airway pressures to a maximum inflation pressure that should not exceed 30 to 35 cmH2O, rather than on strategies that attempt to achieve a normal PaCO2. Because of the decreased overall lung compliance in ARDS patients, the use of low tidal volumes (~ 6 mL/kg of ideal body weight) is usually required. The subsequent decrease in minute ventilation may result in hypercapnia and respiratory acidosis. This ventilatory strategy, which emphasizes the limitation of transpulmonary pressures at the expense of hypercapnia, has been termed permissive hypercapnia.
After intubation, the inspired oxygen fraction (FIO2) is initially set at 1.0 and then decreased in steps to the lowest FIO2 that will maintain an arterial oxygen tension (PaO2) of approximately 60 mmHg. If PaO2 cannot be maintained at 60 mmHg by an FIO2 <0.6, positive end-expiratory pressure (PEEP) may be added (Chap. 266). PEEP improves oxygenation by elevating mean alveolar pressure, thereby recruiting atelectatic alveoli and preventing end-expiratory airway and alveolar closure. In addition, PEEP may prevent alveolar damage by reducing the repetitive and cyclical reopening of closed alveoli during the respiratory cycle. Because PEEP may also overdistend uninvolved alveoli, it should be added cautiously, starting at 5 cmH2O and increasing in increments of 3 to 5 cmH2O to a maximum of 20 to 24 cmH2O. Because airway pressure is transmitted to the pleural space, cardiac output may be adversely affected by the addition of PEEP. In general, the optimal level of PEEP is the amount that achieves an acceptable arterial O2 saturation (<90%) with nontoxic FIO2 levels (<0.6) but without significantly compromising cardiac output. The comprehensive ventilatory strategy that combines low tidal volumes with adequate levels of PEEP has been termed a lung-protective strategy. ARDS patients ventilated with this technique have improved 28-day survival and require less time on mechanical ventilation when compared with ARDS patients treated with conventional ventilation using large tidal volumes achieving normal PaCO2 levels.
Several other ventilatory strategies have been examined with the goal of improving oxygenation. However, none of these techniques has definitively been proven to be beneficial for ARDS patients. When turned from a supine to prone position, ARDS patients develop a more uniform distribution of pleural pressures, with an improvement in ventilation/perfusion matching and better postural drainage of secretions. Prone positioning may improve oxygenation in >75% of ARDS patients. However, the turning of these critically ill patients from the supine to the prone position is not without potential complications, such as unplanned extubation and removal of central venous catheters. The term inverse ratio ventilation is defined when the inspiratory (I) time exceeds the expiratory (E) time (i.e., > one-half of the respiratory cycle; I:E ratio > 1:1). This mode of ventilation is able to maintain a higher mean airway pressure, a major determinant of oxygenation, with lower peak airway pressures than conventional ventilation. However, due to the decrease in expiratory time, inverse ratio ventilation is potentially associated with dynamic hyperinflation and increases in end-expiratory pressure. Finally, partial liquid ventilation with perfluorocarbon, a radiopaque, inert, colorless liquid that carries a large quantity of O2, and CO2, has been studied in patients with severe ARDS. When perfluorocarbon is administered into the trachea of intubated patients, patients can be safely and adequately oxygenated and ventilated with routine mechanical ventilation.
Intravascular Volume Management
Although pulmonary edema in ARDS patients is a consequence of increased permeability of the alveolar-capillary membrane, elevations in the intravascular hydrostatic pressure may also contribute to the accumulation of alveolar liquid and result in worsening oxygenation. Therefore, the optimal fluid management for patients with ARDS requires a balancing between liquid restriction, which may cause hypotension and decreased perfusion to vital organs, and liquid administration, which may increase oxygen requirements. Small decrements in the intravascular volume with diuretic use produce significant decreases in extravascular lung water. Caution must be exercised in reducing intravascular volume, since vigorous diuresis, especially in the setting of PEEP, may reduce cardiac output and perfusion of critical organs. Ideally, the lowest intravascular hydrostatic pressure that also achieves an adequate cardiac output should be maintained. The placement of a pulmonary arterial catheter may be helpful in monitoring cardiac output and pulmonary arterial occlusion pressure (a measure of intravascular volume) in order to optimize the fluid management of patients with ARDS. However, the placement of a pulmonary arterial catheter and the clinical decisions based upon information derived from the catheter do not appear to improve and may actually worsen the outcome of general intensive care unit patients. Therefore the role of the pulmonary arterial catheter for ARDS patients is presently unclear.
Pharmacologic Therapies
Due to their anti-inflammatory properties, glucocorticoids have been used in patients with ARDS, but when administered in high doses (30 mg/kg intravenously every 6 h for a total of four doses), they are not beneficial in the early course of the disease. In contrast, one small randomized study reported an improvement in mortality when glucocorticoids were given after 7 days of unresolving ARDS. In this study, active surveillance for infection was required before enrollment, and glucocorticoids were administered for up to 32 days. Future recommendations regarding the use of these drugs for ARDS patients will be based upon the results of an ongoing multicenter study.
Patients with ARDS have both quantitative and qualitative abnormalities in surfactant, rendering surfactant-replacement therapy an attractive therapeutic modality. In one large randomized study of sepsis-induced ARDS, the administration of synthetic surfactant in an aerosolized form had no significant effect on outcome. Due to concerns with the efficacy of the delivery technique and the lack of essential surfactant-associated proteins in this particular replacement therapy, further studies of different surfactant preparations and modes of administration are presently ongoing.
When inhaled, nitric oxide vasodilates the pulmonary vasculature adjacent to well-ventilated alveoli, thereby improving ventilation-perfusion mismatching. Because of its subsequent inactivation by hemoglobin, nitric oxide produces a selective pulmonary vasodilation without systemic hemodynamic effects. Though inhaled nitric oxide appears to improve oxygenation initially, it is presently unknown whether this therapy will reduce mortality rates in ARDS patients.
Prognosis
Since the initial descriptions of ARDS, mortality rates have ranged from 50 to 70%, although they may now be declining with optimal therapy. Mortality rates are higher in patients over 65 years of age, in those with an at-risk diagnosis of sepsis, and when associated with dysfunction of other organ systems. The cause of death for patients with ARDS has been traditionally divided into early causes (within 72 h) and late causes (after 3 days). Most early deaths are attributed to the original presenting illness or injury. Secondary infection and sepsis, persistent respiratory failure, and multiple-organ dysfunction are the most common causes of death in those ARDS patients who live at least 3 days.
In survivors of ARDS, abnormalities in pulmonary function normally improve considerably by 3 months and reach maximum levels of correction by 6 months after extubation. Although pulmonary function markedly recovers in many survivors, over 50% of these patients will continue to have abnormalities, including restrictive impairment or decreased diffusing capacity. Patients with severe ARDS, characterized by extreme hypoxemia and a longer duration of illness, usually have more pulmonary dysfunction than individuals with mild ARDS. Survivors of ARDS also have significant reductions in their quality of life, specifically in regard to physical functioning when compared to other previously critically ill patients.