Rheumatoid Arthritis
Rheumatoid
arthritis (RA) is a chronic multisystem disease of unknown cause. Although
there are a variety of systemic manifestations, the characteristic feature of
RA is persistent inflammatory synovitis, usually involving peripheral joints in
a symmetric distribution. The potential of the synovial inflammation to cause
cartilage destruction and bone erosions and subsequent changes in joint
integrity is the hallmark of the disease. Despite its destructive potential,
the course of RA can be quite variable. Some patients may experience only a
mild oligoarticular illness of brief duration with minimal joint damage,
whereas others will have a relentless progressive polyarthritis with marked
functional impairment.
Epidemiology and Genetics
The
prevalence of RA is approximately 0.8% of the population (range 0.3 to 2.1%);
women are affected approximately three times more often than men. The
prevalence increases with age, and sex differences diminish in the older age
group. RA is seen throughout the world and affects all races. However, the
incidence and severity seem to be less in rural sub-Saharan Africa and in
Caribbean blacks. The onset is most frequent during the fourth and fifth
decades of life, with 80% of all patients developing the disease between the
ages of 35 and 50. The incidence of RA is more than six times as great in 60-
to 64-year-old women compared to 18- to 29-year-old women.
Family
studies indicate a genetic predisposition. For example, severe RA is found at
approximately four times the expected rate in first-degree relatives of
individuals with disease associated with the presence of the autoantibody,
rheumatoid factor; approximately 10% of patients with RA will have an affected
first-degree relative. Moreover, monozygotic twins are at least four times more
likely to be concordant for RA than dizygotic twins, who have a similar risk of
developing RA as nontwin siblings. Only 15 to 20% of monozygotic twins are
concordant for RA, however, implying that factors other than genetics play an
important etiopathogenic role. Of note, the highest risk for concordance of RA
is noted in twins who have two HLA-DRB1 alleles known to be associated with RA.
The class II major histocompatibility complex allele HLA-DR4. (DRB1*0401) and
related alleles are known to be major genetic risk factors for RA. Early
studies showed that as many as 70% of patients with classic or definite RA
express HLA-DR4 compared with 28% of control individuals. An association with
HLA-DR4 has been noted in many populations, including North American and
European whites, Chippewa Indians, Japanese, and native populations in India,
Mexico, South America, and southern China. In a number of groups, including
Israeli Jews, Asian Indians, and Yakima Indians of North America, however,
there is no association between the development of RA and HLA-DR4. In these
individuals, there is an association between RA and HLA-DR1 in the former two
groups and HLA-Dw16 in the latter. Molecular analysis of HLA-DR antigens has
provided insight into these apparently disparate findings. The HLA-DR molecule
is composed of two chains, a nonpolymorphic ![]() chain
and a highly polymorphic
chain
and a highly polymorphic ![]() chain.
Allelic variations in the HLA-DR molecule reflect differences in the amino
acids of the
chain.
Allelic variations in the HLA-DR molecule reflect differences in the amino
acids of the ![]() chain,
with the major amino acid changes occurring in the three hypervariable regions
of the molecule. Each of the HLA-DR molecules that is associated with RA has
the same or a very similar sequence of amino acids in the third hypervariable
region of the
chain,
with the major amino acid changes occurring in the three hypervariable regions
of the molecule. Each of the HLA-DR molecules that is associated with RA has
the same or a very similar sequence of amino acids in the third hypervariable
region of the ![]() chain
of the molecule. Thus the
chain
of the molecule. Thus the ![]() chains
of the HLA-DR molecules associated with RA, including HLA-Dw4 (DR
chains
of the HLA-DR molecules associated with RA, including HLA-Dw4 (DR![]() 1*0401),
HLA-Dw14 (DR
1*0401),
HLA-Dw14 (DR![]() 1*0404),
HLA-Dw15 (DR
1*0404),
HLA-Dw15 (DR![]() 1*0405),
HLA-DR1 (DR
1*0405),
HLA-DR1 (DR![]() 1*0101),
and HLA-Dw16 (DR
1*0101),
and HLA-Dw16 (DR![]() 1*1402),
contain the same amino acids at positions 67 through 74, with the exception of
a single change of one basic amino acid for another (arginine
1*1402),
contain the same amino acids at positions 67 through 74, with the exception of
a single change of one basic amino acid for another (arginine ![]() lysine)
in position 71 of HLA-Dw4. All other HLA-DR
lysine)
in position 71 of HLA-Dw4. All other HLA-DR ![]() chains
have amino acid changes in this region that alter either their charge or
hydrophobicity. These results indicate that a particular amino acid sequence in
the third hypervariable region of the HLA-DR molecule is a major genetic
element conveying susceptibility to RA, regardless of whether it occurs in
HLA-DR4, HLA-Dw16, or HLA-DR1. It has been estimated that the risk of
developing RA in a person with HLA-Dw4 (DR
chains
have amino acid changes in this region that alter either their charge or
hydrophobicity. These results indicate that a particular amino acid sequence in
the third hypervariable region of the HLA-DR molecule is a major genetic
element conveying susceptibility to RA, regardless of whether it occurs in
HLA-DR4, HLA-Dw16, or HLA-DR1. It has been estimated that the risk of
developing RA in a person with HLA-Dw4 (DR![]() 1*0401)
or HLA-Dw14 (DR
1*0401)
or HLA-Dw14 (DR![]() 1*0404)
is 1 in 35 and 1 in 20, respectively, whereas the presence of both alleles puts
persons at an even greater risk. The lack of association of HLA-DR4 and RA in
certain populations is explained by the major member of the DR4 family found in
that population. HLA-DR4 is a family of closely related, serologically defined
molecules, including HLA-Dw4, -Dw10, -Dw13, and -Dw15. Different members of the
HLA-DR family of molecules are found to predominate in different ethnic groups.
Thus, in HLA-DR4-positive North American whites, HLA-Dw4 and -Dw14 are the most
frequent, whereas HLA-Dw15 is most frequent in Japanese and southern Chinese.
Each of these is associated with RA. By contrast, HLA-Dw10, which is not
associated with RA and contains nonconservative amino acid changes in positions
70 and 71 of the
1*0404)
is 1 in 35 and 1 in 20, respectively, whereas the presence of both alleles puts
persons at an even greater risk. The lack of association of HLA-DR4 and RA in
certain populations is explained by the major member of the DR4 family found in
that population. HLA-DR4 is a family of closely related, serologically defined
molecules, including HLA-Dw4, -Dw10, -Dw13, and -Dw15. Different members of the
HLA-DR family of molecules are found to predominate in different ethnic groups.
Thus, in HLA-DR4-positive North American whites, HLA-Dw4 and -Dw14 are the most
frequent, whereas HLA-Dw15 is most frequent in Japanese and southern Chinese.
Each of these is associated with RA. By contrast, HLA-Dw10, which is not
associated with RA and contains nonconservative amino acid changes in positions
70 and 71 of the ![]() chain,
is most common in Israeli Jews. Therefore, HLA-DR4 is not associated with RA in
this population. In certain groups of patients, there does not appear to be a
clear association between HLA-DR4-related epitopes and RA. Thus, nearly 75% of
African American RA patients do not have this genetic element. Moreover, there
is an association with HLA-DR10 (DRB1*1001) in Spanish and Italian patients,
with HLA-DR9 (DRB1*0901) in Chileans, and with HLA-DR3 (DRB1*0301) in Arab
populations.
chain,
is most common in Israeli Jews. Therefore, HLA-DR4 is not associated with RA in
this population. In certain groups of patients, there does not appear to be a
clear association between HLA-DR4-related epitopes and RA. Thus, nearly 75% of
African American RA patients do not have this genetic element. Moreover, there
is an association with HLA-DR10 (DRB1*1001) in Spanish and Italian patients,
with HLA-DR9 (DRB1*0901) in Chileans, and with HLA-DR3 (DRB1*0301) in Arab
populations.
Additional
genes in the HLA-D complex may also convey altered susceptibility to RA.
Certain HLA-DR alleles, including HLA-DR5 (DRB1*1101), HLA-DR2 (DRB1*1501),
HLA-DR3 (DRB1*0301), and HLA-DR7 (DRB1*0701), may protect against the
development of RA in that they tend to be found at lower frequency in RA
patients than in controls. Moreover, the HLA-DQ alleles, DQB1*0301 and
DQB1*0302, that are in linkage disequilibrium with HLA-DR4 and DQB1*0501, have
also been associated with RA. This has raised the possibility that HLA-DQ
alleles may represent the actual RA susceptibility genes, whereas specific
HLA-DR alleles may convey protection. In this model, the complement of HLA-DR
and DQ alleles determines RA susceptibility. Disease manifestations have also
been associated with HLA phenotype. Thus, early aggressive disease and
extraarticular manifestations are more frequent in patients with DRB1*0401 or
DRB1*0404, and more slowly progressive disease in those with DRB1*0101. The
presence of both DRB1*0401 and DRB1*0404 appears to increase the risk for both
aggressive articular and extraarticular disease. It has been estimated that HLA
genes contribute only a portion of the genetic susceptibility to RA. Thus genes
outside the HLA complex also contribute. These include genes controlling the
expression of the antigen receptor on T cells and both immunoglobulin heavy and
light chains. Moreover, polymorphisms in the tumor necrosis factor (TNF) ![]() and
the interleukin (IL) 10 genes are also associated with RA, as is a region on
chromosome 3 (3q13).
and
the interleukin (IL) 10 genes are also associated with RA, as is a region on
chromosome 3 (3q13).
Genetic
risk factors do not fully account for the incidence of RA, suggesting that
environmental factors also play a role in the etiology of the disease. This is
emphasized by epidemiologic studies in Africa that have indicated that climate
and urbanization have a major impact on the incidence and severity of RA in
groups of similar genetic background.
Etiology
The
cause of RA remains unknown. It has been suggested that RA might be a
manifestation of the response to an infectious agent in a genetically susceptible
host. Because of the worldwide distribution of RA, it has been hypothesized
that if an infectious agent is involved, the organism must be ubiquitous. A
number of possible causative agents have been suggested, including Mycoplasma,
Epstein-Barr virus (EBV), cytomegalovirus, parvovirus, and rubella virus, but
convincing evidence that these or other infectious agents cause RA has not
emerged. The process by which an infectious agent might cause chronic
inflammatory arthritis with a characteristic distribution also remains a matter
of controversy. One possibility is that there is persistent infection of
articular structures or retention of microbial products in the synovial tissues
which generates a chronic inflammatory response. Alternatively, the microorganism
or response to the microorganism might induce an immune response to components
of the joint by altering its integrity and revealing antigenic peptides. In
this regard, reactivity to type II collagen and heat shock proteins has been
demonstrated. Another possibility is that the infecting microorganism might
prime the host to cross-reactive determinants expressed within the joint as a
result of "molecular mimicry." Recent evidence of similarity between
products of certain gram-negative bacteria and EBV and the HLA-DR4 molecule
itself has supported this possibility. Finally, products of infecting
microorganisms might induce the disease. Recent work has focused on the
possible role of "superantigens" produced by a number of
microorganisms, including staphylococci, streptococci and M. arthritidis.
Superantigens are proteins with the capacity to bind to HLA-DR molecules and
particular V![]() segments of the heterodimeric T cell receptor and stimulate specific T cells
expressing the V
segments of the heterodimeric T cell receptor and stimulate specific T cells
expressing the V![]() gene products (Chap. 305). The role of superantigens in the etiology of RA
remains speculative. Of all the potential environmental triggers, the only one
clearly associated with the development of RA is cigarette smoking
gene products (Chap. 305). The role of superantigens in the etiology of RA
remains speculative. Of all the potential environmental triggers, the only one
clearly associated with the development of RA is cigarette smoking
Pathology and Pathogenesis
Microvascular
injury and an increase in the number of synovial lining cells appear to be the
earliest lesions in rheumatoid synovitis. The nature of the insult causing this
response is not known. Subsequently, an increased number of synovial lining
cells is seen along with perivascular infiltration with mononuclear cells.
Before the onset of clinical symptoms, the perivascular infiltrate is
predominantly composed of myeloid cells, whereas in symptomatic arthritis, T
cells can also be found, although their number does not appear to correlate
with symptoms. As the process continues, the synovium becomes edematous and
protrudes into the joint cavity as villous projections.
Light-microscopic
examination discloses a characteristic constellation of features, which include
hyperplasia and hypertrophy of the synovial lining cells; focal or segmental
vascular changes, including microvascular injury, thrombosis, and
neovascularization; edema; and infiltration with mononuclear cells, often
collected into aggregates around small blood vessels (Fig. 312-1). The
endothelial cells of the rheumatoid synovium have the appearance of high
endothelial venules of lymphoid organs and have been altered by cytokine
exposure to facilitate entry of cells into tissue. Rheumatoid synovial endothelial
cells express increased amounts of various adhesion molecules involved in this
process. Although this pathologic picture is typical of RA, it can also be seen
in a variety of other chronic inflammatory arthritides. The mononuclear cell
collections are variable in composition and size. The predominant infiltrating
cell is the T lymphocyte. CD4+ T cells predominate over CD8+ T cells and are
frequently found in close proximity to HLA-DR+ macrophages and dendritic cells.
Increased numbers of a separate population of T cells expressing the ![]()
![]() form
of the T cell receptor have also been found in the synovium, although they
remain a minor population there and their role in RA has not been delineated.
The major population of T cells in the rheumatoid synovium is composed of CD4+
memory T cells that form the majority of cells aggregated around postcapillary
venules. Scattered throughout the tissue are CD8+ T cells. Both populations
express the early activation antigen, CD69. Besides the accumulation of T
cells, rheumatoid synovitis is also characterized by the infiltration of
variable numbers of B cells and antibody-producing plasma cells. In advanced
disease, structures similar to germinal centers of secondary lymphoid organs
may be observed in the synovium. Both polyclonal immunoglobulin and the
autoantibody rheumatoid factor are produced within the synovial tissue, which
leads to the local formation of immune complexes. Finally, the synovial
fibroblasts in RA manifest evidence of activation in that they produce a number
of enzymes such as collagenase and cathepsins that can degrade components of
the articular matrix. These activated fibroblasts are particularly prominent in
the lining layer and at the interface with bone and cartilage. Osteoclasts are
also prominent at sites of bone erosion.
form
of the T cell receptor have also been found in the synovium, although they
remain a minor population there and their role in RA has not been delineated.
The major population of T cells in the rheumatoid synovium is composed of CD4+
memory T cells that form the majority of cells aggregated around postcapillary
venules. Scattered throughout the tissue are CD8+ T cells. Both populations
express the early activation antigen, CD69. Besides the accumulation of T
cells, rheumatoid synovitis is also characterized by the infiltration of
variable numbers of B cells and antibody-producing plasma cells. In advanced
disease, structures similar to germinal centers of secondary lymphoid organs
may be observed in the synovium. Both polyclonal immunoglobulin and the
autoantibody rheumatoid factor are produced within the synovial tissue, which
leads to the local formation of immune complexes. Finally, the synovial
fibroblasts in RA manifest evidence of activation in that they produce a number
of enzymes such as collagenase and cathepsins that can degrade components of
the articular matrix. These activated fibroblasts are particularly prominent in
the lining layer and at the interface with bone and cartilage. Osteoclasts are
also prominent at sites of bone erosion.
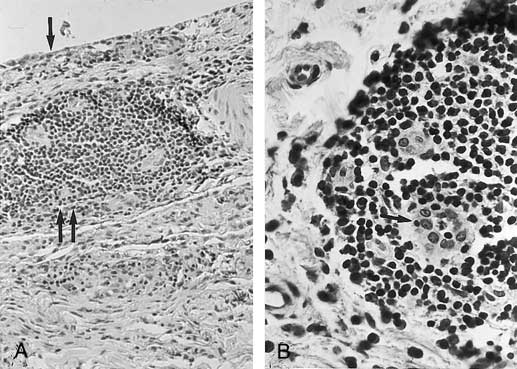
Figure 312-1: Histology of rheumatoid synovitis. A.
The characteristic features of rheumatoid inflammation with hyperplasia of the
lining layer (arrow) and mononuclear infiltrates in the sublining layer (double
arrow). B. A higher magnification of the largely CD4+ T cell
infiltrate around postcapillary venules (arrow).
The
rheumatoid synovium is characterized by the presence of a number of secreted
products of activated lymphocytes, macrophages, and fibroblasts. The local
production of these cytokines and chemokines appears to account for many of the
pathologic and clinical manifestations of RA. These effector molecules include
those that are derived from T lymphocytes such as interleukin IL-2, interferon
(IFN) ![]() ,
IL-6, IL-10, granulocyte-macrophage colony stimulating factor (GM-CSF), TNF-
,
IL-6, IL-10, granulocyte-macrophage colony stimulating factor (GM-CSF), TNF-![]() ,
transforming growth factor
,
transforming growth factor ![]() (TGF-
(TGF-![]() );
IL-13, IL-16, and IL-17; those originating from activated myeloid cells,
including IL-1, TNF-
);
IL-13, IL-16, and IL-17; those originating from activated myeloid cells,
including IL-1, TNF-![]() ,
IL-6, IL-8, IL-10, IL-12, GM-CSF, macrophage CSF, platelet-derived growth
factor, insulin-like growth factor, and TGF-
,
IL-6, IL-8, IL-10, IL-12, GM-CSF, macrophage CSF, platelet-derived growth
factor, insulin-like growth factor, and TGF-![]() ;
as well as those secreted by other cell types in the synovium, such as
fibroblasts and endothelial cells, including IL-1, IL-6, IL-8, GM-CSF, IL-15,
IL-16, IL-18, and macrophage CSF. The activity of these chemokines and
cytokines appears to account for many of the features of rheumatoid synovitis,
including the synovial tissue inflammation, synovial fluid inflammation,
synovial proliferation, and cartilage and bone damage, as well as the systemic
manifestations of RA. In addition to the production of effector molecules that
propagate the inflammatory process, local factors are produced that tend to
slow the inflammation, including specific inhibitors of cytokine action and
additional cytokines, such as TGF-
;
as well as those secreted by other cell types in the synovium, such as
fibroblasts and endothelial cells, including IL-1, IL-6, IL-8, GM-CSF, IL-15,
IL-16, IL-18, and macrophage CSF. The activity of these chemokines and
cytokines appears to account for many of the features of rheumatoid synovitis,
including the synovial tissue inflammation, synovial fluid inflammation,
synovial proliferation, and cartilage and bone damage, as well as the systemic
manifestations of RA. In addition to the production of effector molecules that
propagate the inflammatory process, local factors are produced that tend to
slow the inflammation, including specific inhibitors of cytokine action and
additional cytokines, such as TGF-![]() ,
which inhibits many of the features of rheumatoid synovitis including T cell
activation and proliferation, B cell differentiation, and migration of cells
into the inflammatory site.
,
which inhibits many of the features of rheumatoid synovitis including T cell
activation and proliferation, B cell differentiation, and migration of cells
into the inflammatory site.
These
findings have suggested that the propagation of RA is an immunologically
mediated event, although the original initiating stimulus has not been
characterized. One view is that the inflammatory process in the tissue is
driven by the CD4+ T cells infiltrating the synovium. Evidence for this
includes (1) the predominance of CD4+ T cells in the synovium; (2) the increase
in soluble IL-2 receptors, a product of activated T cells, in blood and
synovial fluid of patients with active RA; and (3) amelioration of the disease
by removal of T cells by thoracic duct drainage or peripheral lymphapheresis or
suppression of their function by drugs, such as cyclosporine or nondepleting
monoclonal antibodies to CD4. In addition, the association of RA with certain
HLA-DR or -DQ alleles, whose only known functions are to shape the repertoire
of CD4+ T cells during ontogeny in the thymus and bind and present antigenic
peptides to CD4+ T cells in the periphery, strongly implies a role for CD4+ T
cells in the pathogenesis of the disease. Finally, patients with established RA
who become infected with HIV also have been noted to improve, although this has
not been a uniform finding. Within the rheumatoid synovium, the CD4+ T cells
differentiate predominantly into Th1-like effector cells producing the
proinflammatory cytokine IFN-![]() and appear to be deficient in differentiation into Th2-like effector cells
capable of producing the anti-inflammatory cytokine IL-4. As a result of the
ongoing secretion of IFN-
and appear to be deficient in differentiation into Th2-like effector cells
capable of producing the anti-inflammatory cytokine IL-4. As a result of the
ongoing secretion of IFN-![]() without the regulatory influences of IL-4, macrophages are activated to produce
the proinflammatory cytokines IL-1 and TNF-
without the regulatory influences of IL-4, macrophages are activated to produce
the proinflammatory cytokines IL-1 and TNF-![]() and also increase expression of HLA molecules. Moreover, T lymphocytes express
surface molecules such as CD154 (CD40 ligand) and also produce a variety of
cytokines that promote B cell proliferation and differentiation into antibody-forming
cells and therefore also may promote local B cell stimulation. The resultant
production of immunoglobulin and rheumatoid factor can lead to immune-complex
formation with consequent complement activation and exacerbation of the
inflammatory process by the production of the anaphylatoxins, C3a and C5a, and
the chemotactic factor C5a. The tissue inflammation is reminiscent of delayed
type hypersensitivity reactions occurring in response to soluble antigens or
microorganisms, although it has become clear that the number of T cells
producing cytokines such as IFN-
and also increase expression of HLA molecules. Moreover, T lymphocytes express
surface molecules such as CD154 (CD40 ligand) and also produce a variety of
cytokines that promote B cell proliferation and differentiation into antibody-forming
cells and therefore also may promote local B cell stimulation. The resultant
production of immunoglobulin and rheumatoid factor can lead to immune-complex
formation with consequent complement activation and exacerbation of the
inflammatory process by the production of the anaphylatoxins, C3a and C5a, and
the chemotactic factor C5a. The tissue inflammation is reminiscent of delayed
type hypersensitivity reactions occurring in response to soluble antigens or
microorganisms, although it has become clear that the number of T cells
producing cytokines such as IFN-![]() is less than is found in typical delayed type hypersensitivity reactions,
perhaps owing to the large amount of reactive oxygen species produced locally
in the synovium that can dampen T cell function. It remains unclear whether the
persistent T cell activity represents a response to a persistent exogenous antigen
or to altered autoantigens such as collagen, immunoglobulin, or one of the heat
shock proteins, or perhaps both. Alternatively, it could represent persistent
responsiveness to activated autologous cells such as might occur as a result of
EBV infection or persistent response to a foreign antigen or superantigen in
the synovial tissue. Finally, rheumatoid inflammation could reflect persistent
stimulation of T cells by synovial-derived antigens that cross-react with
determinants introduced during antecedent exposure to foreign antigens or
infectious microorganisms.
is less than is found in typical delayed type hypersensitivity reactions,
perhaps owing to the large amount of reactive oxygen species produced locally
in the synovium that can dampen T cell function. It remains unclear whether the
persistent T cell activity represents a response to a persistent exogenous antigen
or to altered autoantigens such as collagen, immunoglobulin, or one of the heat
shock proteins, or perhaps both. Alternatively, it could represent persistent
responsiveness to activated autologous cells such as might occur as a result of
EBV infection or persistent response to a foreign antigen or superantigen in
the synovial tissue. Finally, rheumatoid inflammation could reflect persistent
stimulation of T cells by synovial-derived antigens that cross-react with
determinants introduced during antecedent exposure to foreign antigens or
infectious microorganisms.
Overriding
the chronic inflammation in the synovial tissue is an acute inflammatory
process in the synovial fluid. The exudative synovial fluid contains more
polymorphonuclear leukocytes than mononuclear cells. A number of mechanisms
play a role in stimulating the exudation of synovial fluid. Locally produced
immune complexes can activate complement and generate anaphylatoxins and
chemotactic factors. Local production, by a variety of cells, of chemokines and
cytokines with chemotactic activity as well as inflammatory mediators such as
leukotriene B4 and products of complement activation can attract
neutrophils. Moreover, many of these same agents can also stimulate the
endothelial cells of postcapillary venules to become more efficient at binding
circulating cells. The net result is the enhanced migration of
polymorphonuclear leukocytes into the synovial site. In addition, vasoactive
mediators such as histamine produced by the mast cells that infiltrate the
rheumatoid synovium may also facilitate the exudation of inflammatory cells
into the synovial fluid. Finally, the vasodilatory effects of locally produced
prostaglandin E2 may also facilitate entry of inflammatory cells
into the inflammatory site. Once in the synovial fluid, the polymorphonuclear
leukocytes can ingest immune complexes, with the resultant production of
reactive oxygen metabolites and other inflammatory mediators, further adding to
the inflammatory milieu. Locally produced cytokines and chemokines can
additionally stimulate polymorphonuclear leukocytes. The production of large
amounts of cyclooxygenase and lipoxygenase pathway products of arachidonic acid
metabolism by cells in the synovial fluid and tissue further accentuates the
signs and symptoms of inflammation.
The
precise mechanism by which bone and cartilage destruction occurs has not been
completely resolved. Although the synovial fluid contains a number of enzymes
potentially able to degrade cartilage, the majority of destruction occurs in
juxtaposition to the inflamed synovium, or pannus, that spreads to cover the
articular cartilage. This vascular granulation tissue is composed of
proliferating fibroblasts, small blood vessels, and a variable number of
mononuclear cells and produces a large amount of degradative enzymes, including
collagenase and stromelysin, that may facilitate tissue damage. The cytokines
IL-1 and TNF-![]() play an important role by stimulating the cells of the pannus to produce
collagenase and other neutral proteases. These same two cytokines also activate
chondrocytes in situ, stimulating them to produce proteolytic enzymes that can
degrade cartilage locally and also inhibiting synthesis of new matrix
molecules. Finally, these two cytokines may contribute to the local
demineralization of bone by activating osteoclasts that accumulate at the site
of local bone resorption. Prostaglandin E2 produced by fibroblasts
and macrophages may also contribute to bone demineralization. The common final
pathway of bone erosion is likely to involve the activation of osteoclasts that
are present in large numbers at these sites. Systemic manifestations of RA can
be accounted for by release of inflammatory effector molecules from the
synovium. These include IL-1, TNF-
play an important role by stimulating the cells of the pannus to produce
collagenase and other neutral proteases. These same two cytokines also activate
chondrocytes in situ, stimulating them to produce proteolytic enzymes that can
degrade cartilage locally and also inhibiting synthesis of new matrix
molecules. Finally, these two cytokines may contribute to the local
demineralization of bone by activating osteoclasts that accumulate at the site
of local bone resorption. Prostaglandin E2 produced by fibroblasts
and macrophages may also contribute to bone demineralization. The common final
pathway of bone erosion is likely to involve the activation of osteoclasts that
are present in large numbers at these sites. Systemic manifestations of RA can
be accounted for by release of inflammatory effector molecules from the
synovium. These include IL-1, TNF-![]() ,
and IL-6, which account for many of the manifestations of active RA, including
malaise, fatigue, and elevated levels of serum acute-phase reactants. The
importance of TNF-
,
and IL-6, which account for many of the manifestations of active RA, including
malaise, fatigue, and elevated levels of serum acute-phase reactants. The
importance of TNF-![]() in producing these manifestations is emphasized by the prompt amelioration of
symptoms following administration of a monoclonal antibody to TNF-
in producing these manifestations is emphasized by the prompt amelioration of
symptoms following administration of a monoclonal antibody to TNF-![]() or a soluble TNF-
or a soluble TNF-![]() receptor Ig construct to patients with RA. In addition, immune complexes
produced within the synovium and entering the circulation may account for other
features of the disease, such as systemic vasculitis.
receptor Ig construct to patients with RA. In addition, immune complexes
produced within the synovium and entering the circulation may account for other
features of the disease, such as systemic vasculitis.
As
shown in Fig. 312-2, the pathology of RA evolves over the duration of this
chronic disease. The earliest event appears to be a nonspecific inflammatory
response initiated by an unknown stimulus and characterized by accumulation of
macrophages and other mononuclear cells within the sublining layer of the
synovium. The activity of these cells is demonstrated by the increased
appearance of macrophage-derived cytokines, including TNF-![]() ,
IL-1
,
IL-1![]() ,
and IL-6. Subsequently, activation of CD4+ T cells is induced, presumably in
response to antigenic peptides displayed by a variety of antigen-presenting
cells in the synovial tissue. The activated T cells are capable of producing
cytokines, especially IFN-
,
and IL-6. Subsequently, activation of CD4+ T cells is induced, presumably in
response to antigenic peptides displayed by a variety of antigen-presenting
cells in the synovial tissue. The activated T cells are capable of producing
cytokines, especially IFN-![]() ,
which amplify and perpetuate the inflammation. The presence of activated T
cells expressing CD154 (CD40 ligand) can induce polyclonal B cell stimulation
and the local production of rheumatoid factor. The cascade of cytokines
produced in the synovium activates a variety of cells in the synovium, bone,
and cartilage to produce effector molecules that can cause tissue damage
characteristic of chronic inflammation. It is important to emphasize that there
is no current way to predict the progress from one stage of inflammation to the
next, and once established, each can influence the other. Important features of
this model include the following: (1) the major pathologic events vary with
time in this chronic disease; (2) the time required to progress from one step
to the next may vary in different patients and the events, once established,
may persist simultaneously; (3) once established, the major pathogenic events
operative in an individual patient may vary at different times; and (4) the
process is chronic and reiterative, with successive events stimulating
progressive amplification of inflammation. These considerations have important
implications with regard to appropriate treatment.
,
which amplify and perpetuate the inflammation. The presence of activated T
cells expressing CD154 (CD40 ligand) can induce polyclonal B cell stimulation
and the local production of rheumatoid factor. The cascade of cytokines
produced in the synovium activates a variety of cells in the synovium, bone,
and cartilage to produce effector molecules that can cause tissue damage
characteristic of chronic inflammation. It is important to emphasize that there
is no current way to predict the progress from one stage of inflammation to the
next, and once established, each can influence the other. Important features of
this model include the following: (1) the major pathologic events vary with
time in this chronic disease; (2) the time required to progress from one step
to the next may vary in different patients and the events, once established,
may persist simultaneously; (3) once established, the major pathogenic events
operative in an individual patient may vary at different times; and (4) the
process is chronic and reiterative, with successive events stimulating
progressive amplification of inflammation. These considerations have important
implications with regard to appropriate treatment.
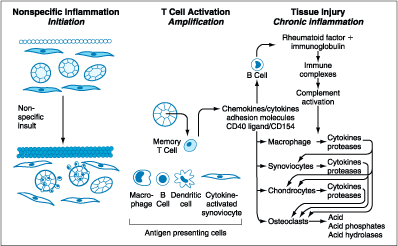
Figure 312-2: The progression of rheumatoid synovitis.
This figure depicts the evolution of the pathogenic mechanisms and ultimate
pathologic changes involved in the development of rheumatoid synovitis. The
stages of rheumatoid arthritis are proposed to be an initiation phase of
nonspecific inflammation, followed by an amplification phase resulting from T
cell activation, and finally a stage of chronic inflammation with tissue
injury. A variety of stimuli may initiate the initial phase of nonspecific
inflammation, which may last for a protracted period of time with no or
moderate symptoms. When activation of memory T cells in response to a variety
of peptides presented by antigen-presenting cells occurs in genetically
susceptible individuals, amplification of inflammation occurs with the
promotion of local rheumatoid factor production and enhanced capacity to
mediate tissue damage.
Clinical Manifestations
Onset
Characteristically,
RA is a chronic polyarthritis. In approximately two-thirds of patients, it
begins insidiously with fatigue, anorexia, generalized weakness, and vague
musculoskeletal symptoms until the appearance of synovitis becomes apparent.
This prodrome may persist for weeks or months and defy diagnosis. Specific
symptoms usually appear gradually as several joints, especially those of the
hands, wrists, knees, and feet, become affected in a symmetric fashion. In
approximately 10% of individuals, the onset is more acute, with a rapid
development of polyarthritis, often accompanied by constitutional symptoms,
including fever, lymphadenopathy, and splenomegaly. In approximately one-third
of patients, symptoms may initially be confined to one or a few joints. Although
the pattern of joint involvement may remain asymmetric in a few patients, a
symmetric pattern is more typical.
Signs and Symptoms of Articular Disease
Pain,
swelling, and tenderness may initially be poorly localized to the joints. Pain
in affected joints, aggravated by movement, is the most common manifestation of
established RA. It corresponds in pattern to the joint involvement but does not
always correlate with the degree of apparent inflammation. Generalized
stiffness is frequent and is usually greatest after periods of inactivity.
Morning stiffness of greater than 1-h duration is an almost invariable feature
of inflammatory arthritis and may serve to distinguish it from various
noninflammatory joint disorders. Notably, however, the presence of morning
stiffness may not reliably distinguish between chronic inflammatory and
noninflammatory arthritides, as it is also found frequently in the latter. The
majority of patients will experience constitutional symptoms such as weakness,
easy fatigability, anorexia, and weight loss. Although fever to 40°C occurs on
occasion, temperature elevation in excess of 38°C is unusual and suggests the
presence of an intercurrent problem such as infection.
Clinically,
synovial inflammation causes swelling, tenderness, and limitation of motion.
Warmth is usually evident on examination, especially of large joints such as
the knee, but erythema is infrequent. Pain originates predominantly from the
joint capsule, which is abundantly supplied with pain fibers and is markedly sensitive
to stretching or distention. Joint swelling results from accumulation of
synovial fluid, hypertrophy of the synovium, and thickening of the joint
capsule. Initially, motion is limited by pain. The inflamed joint is usually
held in flexion to maximize joint volume and minimize distention of the
capsule. Later, fibrous or bony ankylosis or soft tissue contractures lead to
fixed deformities.
Although
inflammation can affect any diarthrodial joint, RA most often causes symmetric
arthritis with characteristic involvement of certain specific joints such as
the proximal interphalangeal and metacarpophalangeal joints. The distal
interphalangeal joints are rarely involved. Synovitis of the wrist joints is a
nearly uniform feature of RA and may lead to limitation of motion, deformity,
and median nerve entrapment (carpal tunnel syndrome). Synovitis of the elbow
joint often leads to flexion contractures that may develop early in the
disease. The knee joint is commonly involved with synovial hypertrophy, chronic
effusion, and frequently ligamentous laxity. Pain and swelling behind the knee
may be caused by extension of inflamed synovium into the popliteal space
(Baker's cyst). Arthritis in the forefoot, ankles, and subtalar joints can
produce severe pain with ambulation as well as a number of deformities. Axial
involvement is usually limited to the upper cervical spine. Involvement of the
lumbar spine is not seen, and lower back pain cannot be ascribed to rheumatoid
inflammation. On occasion, inflammation from the synovial joints and bursae of
the upper cervical spine leads to atlantoaxial subluxation. This usually
presents as pain in the occiput but on rare occasions may lead to compression
of the spinal cord.
With
persistent inflammation, a variety of characteristic joint changes develop.
These can be attributed to a number of pathologic events, including laxity of
supporting soft tissue structures; damage or weakening of ligaments, tendons,
and the joint capsule; cartilage degradation; muscle imbalance; and unopposed
physical forces associated with the use of affected joints. Characteristic
changes of the hand include (1) radial deviation at the wrist with ulnar
deviation of the digits, often with palmar subluxation of the proximal
phalanges ("Z" deformity); (2) hyperextension of the proximal
interphalangeal joints, with compensatory flexion of the distal interphalangeal
joints (swan-neck deformity); (3) flexion contracture of the proximal
interphalangeal joints and extension of the distal interphalangeal joints (boutonnière
deformity); and (4) hyperextension of the first interphalangeal joint and
flexion of the first metacarpophalangeal joint with a consequent loss of thumb
mobility and pinch. Typical joint changes may also develop in the feet,
including eversion at the hindfoot (subtalar joint), plantar subluxation of the
metatarsal heads, widening of the forefoot, hallux valgus, and lateral
deviation and dorsal subluxation of the toes.
Extraarticular Manifestations
RA
is a systemic disease with a variety of extraarticular manifestations. Although
these occur frequently, not all of them have clinical significance. However, on
occasion, they may be the major evidence of disease activity and source of
morbidity and require management per se. As a rule, these manifestations occur
in individuals with high titers of autoantibodies to the Fc component of
immunoglobulin G (rheumatoid factors).
Rheumatoid nodules develop in 20 to 30% of persons with RA. They
are usually found on periarticular structures, extensor surfaces, or other
areas subjected to mechanical pressure, but they can develop elsewhere,
including the pleura and meninges. Common locations include the olecranon
bursa, the proximal ulna, the Achilles tendon, and the occiput. Nodules vary in
size and consistency and are rarely symptomatic, but on occasion they break
down as a result of trauma or become infected. They are found almost invariably
in individuals with circulating rheumatoid factor. Histologically, rheumatoid
nodules consist of a central zone of necrotic material including collagen
fibrils, noncollagenous filaments, and cellular debris; a midzone of palisading
macrophages that express HLA-DR antigens; and an outer zone of granulation
tissue. Examination of early nodules has suggested that the initial event may
be a focal vasculitis. In some patients, treatment with methotrexate can
increase the number of nodules dramatically.
Clinical
weakness and atrophy of skeletal muscle are common. Muscle atrophy may be
evident within weeks of the onset of RA and is usually most apparent in
musculature approximating affected joints. Muscle biopsy may show type II fiber
atrophy and muscle fiber necrosis with or without a mononuclear cell
infiltrate.
Rheumatoid vasculitis (Chap. 317), which can affect nearly any organ
system, is seen in patients with severe RA and high titers of circulating
rheumatoid factor. Rheumatoid vasculitis is very uncommon in African Americans.
In its most aggressive form, rheumatoid vasculitis can cause polyneuropathy and
mononeuritis multiplex, cutaneous ulceration and dermal necrosis, digital
gangrene, and visceral infarction. While such widespread vasculitis is very
rare, more limited forms are not uncommon, especially in white patients with
high titers of rheumatoid factor. Neurovascular disease presenting either as a
mild distal sensory neuropathy or as mononeuritis multiplex may be the only
sign of vasculitis. Cutaneous vasculitis usually presents as crops of small
brown spots in the nail beds, nail folds, and digital pulp. Larger ischemic ulcers,
especially in the lower extremity, may also develop. Myocardial infarction
secondary to rheumatoid vasculitis has been reported, as has vasculitic
involvement of lungs, bowel, liver, spleen, pancreas, lymph nodes, and testes.
Renal vasculitis is rare.
Pleuropulmonary manifestations, which are more
commonly observed in men, include pleural disease, interstitial fibrosis,
pleuropulmonary nodules, pneumonitis, and arteritis. Evidence of pleuritis is
found commonly at autopsy, but symptomatic disease during life is infrequent.
Typically, the pleural fluid contains very low levels of glucose in the absence
of infection. Pleural fluid complement is also low compared with the serum
level when these are related to the total protein concentration. Pulmonary fibrosis
can produce impairment of the diffusing capacity of the lung. Pulmonary nodules
may appear singly or in clusters. When they appear in individuals with
pneumoconiosis, a diffuse nodular fibrotic process (Caplan's syndrome) may
develop. On occasion, pulmonary nodules may cavitate and produce a pneumothorax
or bronchopleural fistula. Rarely, pulmonary hypertension secondary to
obliteration of the pulmonary vasculature occurs. In addition to
pleuropulmonary disease, upper airway obstruction from cricoarytenoid arthritis
or laryngeal nodules may develop.
Clinically
apparent heart disease attributed to the rheumatoid process is rare, but
evidence of asymptomatic pericarditis is found at autopsy in 50% of cases.
Pericardial fluid has a low glucose level and is frequently associated with the
occurrence of pleural effusion. Although pericarditis is usually asymptomatic,
on rare occasions death has occurred from tamponade. Chronic constrictive
pericarditis may also occur.
RA
tends to spare the central nervous system directly, although vasculitis can
cause peripheral neuropathy. Neurologic manifestations may also result
from atlantoaxial or midcervical spine subluxations. Nerve entrapment secondary
to proliferative synovitis or joint deformities may produce neuropathies of
median, ulnar, radial (interosseous branch), or anterior tibial nerves.
The
rheumatoid process involves the eye in fewer than 1% of patients.
Affected individuals usually have long-standing disease and nodules. The two
principal manifestations are episcleritis, which is usually mild and transient,
and scleritis, which involves the deeper layers of the eye and is a more
serious inflammatory condition. Histologically, the lesion is similar to a
rheumatoid nodule and may result in thinning and perforation of the globe
(scleromalacia perforans). From 15 to 20% of persons with RA may develop
Sjögren's syndrome with attendant keratoconjunctivitis sicca.
Felty's syndrome consists of chronic RA, splenomegaly,
neutropenia, and, on occasion, anemia and thrombocytopenia. It is most common
in individuals with long-standing disease. These patients frequently have high
titers of rheumatoid factor, subcutaneous nodules, and other manifestations of
systemic rheumatoid disease. Felty's syndrome is very uncommon in African
Americans. It may develop after joint inflammation has regressed. Circulating
immune complexes are often present, and evidence of complement consumption may
be seen. The leukopenia is a selective neutropenia with polymorphonuclear
leukocyte counts of <1500 cells per microliter and sometimes <1000 cells
per microliter. Bone marrow examination usually reveals moderate
hypercellularity with a paucity of mature neutrophils. However, the bone marrow
may be normal, hyperactive, or hypoactive; maturation arrest may be seen.
Hypersplenism has been proposed as one of the causes of leukopenia, but
splenomegaly is not invariably found and splenectomy does not always correct
the abnormality. Excessive margination of granulocytes caused by antibodies to
these cells, complement activation, or binding of immune complexes may
contribute to granulocytopenia. Patients with Felty's syndrome have increased
frequency of infections usually associated with neutropenia. The cause of the
increased susceptibility to infection is related to the defective function of
polymorphonuclear leukocytes as well as the decreased number of cells.
Osteoporosis secondary to rheumatoid involvement is common
and may be aggravated by glucocorticoid therapy. Glucocorticoid treatment may
cause significant loss of bone mass, especially early in the course of therapy,
even when low doses are employed. Osteopenia in RA involves both juxtaarticular
bone and long bones distant from involved joints. RA is associated with a
modest decrease in mean bone mass and a moderate increase in the risk of
fracture. Bone mass appears to be adversely affected by functional impairment
and active inflammation, especially early in the course of the disease.
RA in the Elderly
The
incidence of RA continues to increase past age 60. It has been suggested that
elderly-onset RA might have a poorer prognosis, as manifested by more
persistent disease activity, more frequent radiographically evident
deterioration, more frequent systemic involvement, and more rapid functional
decline. Aggressive disease is largely restricted to those patients with high
titers of rheumatoid factor. By contrast, elderly patients who develop RA
without elevated titers of rheumatoid factor (seronegative disease) generally
have less severe, often self-limited disease.
Laboratory Findings
No
tests are specific for diagnosing RA. However, rheumatoid factors, which are
autoantibodies reactive with the Fc portion of IgG, are found in more than
two-thirds of adults with the disease. Widely utilized tests largely detect IgM
rheumatoid factors. The presence of rheumatoid factor is not specific for RA.
Rheumatoid factor is found in 5% of healthy persons. The frequency of
rheumatoid factor in the general population increases with age, and 10 to 20%
of individuals over 65 years old have a positive test. In addition, a number of
conditions besides RA are associated with the presence of rheumatoid factor.
These include systemic lupus erythematosus, Sjögren's syndrome, chronic liver
disease, sarcoidosis, interstitial pulmonary fibrosis, infectious
mononucleosis, hepatitis B, tuberculosis, leprosy, syphilis, subacute bacterial
endocarditis, visceral leishmaniasis, schistosomiasis, and malaria. In
addition, rheumatoid factor may appear transiently in normal individuals after
vaccination or transfusion and may also be found in relatives of individuals
with RA.
The
presence of rheumatoid factor does not establish the diagnosis of RA as the
predictive value of the presence of rheumatoid factor in determining a
diagnosis of RA is poor. Thus fewer than one-third of unselected patients with
a positive test for rheumatoid factor will be found to have RA. Therefore, the
rheumatoid factor test is not useful as a screening procedure. However, the
presence of rheumatoid factor can be of prognostic significance because
patients with high titers tend to have more severe and progressive disease with
extraarticular manifestations. Rheumatoid factor is uniformly found in patients
with nodules or vasculitis. In summary, a test for the presence of rheumatoid
factor can be employed to confirm a diagnosis in individuals with a suggestive
clinical presentation and, if present in high titer, to designate patients at
risk for severe systemic disease. A number of additional autoantibodies may be
found in patients with RA, including antibodies to filaggrin, citrulline,
calpastatin, components of the spliceosome (RA-33), and an unknown antigen, Sa.
Some of these may be useful in diagnosis in that they may occur early in the
disease before rheumatoid factor is present or may be associated with
aggressive disease.
Normochromic,
normocytic anemia is frequently present in active RA. It is thought to reflect
ineffective erythropoiesis; large stores of iron are found in the bone marrow.
In general, anemia and thrombocytosis correlate with disease activity. The
white blood cell count is usually normal, but a mild leukocytosis may be
present. Leukopenia may also exist without the full-blown picture of Felty's
syndrome. Eosinophilia, when present, usually reflects severe systemic disease.
The
erythrocyte sedimentation rate is increased in nearly all patients with active
RA. The levels of a variety of other acute-phase reactants including
ceruloplasmin and C-reactive protein are also elevated, and generally such
elevations correlate with disease activity and the likelihood of progressive
joint damage.
Synovial
fluid analysis confirms the presence of inflammatory arthritis, although none
of the findings is specific. The fluid is usually turbid, with reduced
viscosity, increased protein content, and a slightly decreased or normal
glucose concentration. The white cell count varies between 5 and 50,000/![]() L;
polymorphonuclear leukocytes predominate. A synovial fluid white blood cell
count >2000/
L;
polymorphonuclear leukocytes predominate. A synovial fluid white blood cell
count >2000/![]() L
with more than 75% polymorphonuclear leukocytes is highly characteristic of
inflammatory arthritis, although not diagnostic of RA. Total hemolytic
complement, C3, and C4 are markedly diminished in synovial fluid relative to
total protein concentration as a result of activation of the classic complement
pathway by locally produced immune complexes.
L
with more than 75% polymorphonuclear leukocytes is highly characteristic of
inflammatory arthritis, although not diagnostic of RA. Total hemolytic
complement, C3, and C4 are markedly diminished in synovial fluid relative to
total protein concentration as a result of activation of the classic complement
pathway by locally produced immune complexes.
Radiographic Evaluation
Early
in the disease, roentgenograms of the affected joints are usually not helpful
in establishing a diagnosis. They reveal only that which is apparent from
physical examination, namely, evidence of soft tissue swelling and joint
effusion. As the disease progresses, abnormalities become more pronounced, but
none of the radiographic findings is diagnostic of RA. The diagnosis, however,
is supported by a characteristic pattern of abnormalities, including the
tendency toward symmetric involvement. Juxtaarticular osteopenia may become
apparent within weeks of onset. Loss of articular cartilage and bone erosions
develop after months of sustained activity. The primary value of radiography is
to determine the extent of cartilage destruction and bone erosion produced by
the disease, particularly when one is monitoring the impact of therapy with
disease-modifying drugs or surgical intervention. Other means of imaging bones
and joints, including 99mTc bisphosphonate bone scanning and
magnetic resonance imaging, may be capable of detecting early inflammatory
changes that are not apparent from standard radiography but are rarely
necessary in the routine evaluation of patients with RA.
Clinical Course and Prognosis
The
course of RA is quite variable and difficult to predict in an individual
patient. Most patients experience persistent but fluctuating disease activity,
accompanied by a variable degree of joint abnormalities and functional
impairment. After 10 to 12 years, fewer than 20% of patients will have no
evidence of disability or joint abnormalities. Within 10 years, approximately
50% of patients will have work disability. A number of features are correlated
with a greater likelihood of developing joint abnormalities or disabilities.
These include the presence of more than 20 inflamed joints, a markedly elevated
erythrocyte sedimentation rate, radiographic evidence of bone erosions, the
presence of rheumatoid nodules, high titers of serum rheumatoid factor, the
presence of functional disability, persistent inflammation, advanced age at
onset, the presence of comorbid conditions, low socioeconomic status or
educational level, or the presence of HLA-DRB1*0401 or -DRB*0404. The presence
of one or more of these implies the presence of more aggressive disease with a
greater likelihood of developing progressive joint abnormalities and
disability. Persistent elevation of the erythrocyte sedimentation rate,
disability, and pain on longitudinal follow-up are good predictors of work
disability. Patients who lack these features have more indolent disease with a
slower progression to joint abnormalities and disability. The pattern of
disease onset does not appear to predict the development of disabilities.
Approximately 15% of patients with RA will have a short-lived inflammatory
process that remits without major disability. These individuals tend to lack
the aforementioned features associated with more aggressive disease.
Several
features of patients with RA appear to have prognostic significance. Remissions
of disease activity are most likely to occur during the first year. White
females tend to have more persistent synovitis and more progressively erosive
disease than males. Persons who present with high titers of rheumatoid factor,
C-reactive protein, and haptoglobin also have a worse prognosis, as do
individuals with subcutaneous nodules or radiographic evidence of erosions at
the time of initial evaluation. Sustained disease activity of more than 1
year's duration portends a poor outcome, and persistent elevation of
acute-phase reactants appears to correlate strongly with radiographic
progression. A large proportion of inflamed joints manifest erosions within 2
years, whereas the subsequent course of erosions is highly variable; however,
in general, radiographic damage appears to progress at a constant rate in
patients with RA. Foot joints are affected more frequently than hand joints.
Despite the decrease in the rate of progressive joint damage with time,
functional disability, which develops early in the course of the disease,
continues to worsen at the same rate, although the most rapid rate of
functional loss occurs within the first 2 years of disease.
The
median life expectancy of persons with RA is shortened by 3 to 7 years. Of the
2.5-fold increase in mortality rate, RA itself is a contributing feature in 15
to 30%. The increased mortality rate seems to be limited to patients with more
severe articular disease and can be attributed largely to infection and
gastrointestinal bleeding. Drug therapy may also play a role in the increased
mortality rate seen in these individuals. Factors correlated with early death
include disability, disease duration or severity, glucocorticoid use, age at
onset, and low socioeconomic or educational status.
Diagnosis
The
mean delay from disease onset to diagnosis is 9 months. This is often related
to the nonspecific nature of initial symptoms. The diagnosis of RA is easily
made in persons with typical established disease. In a majority of patients,
the disease assumes its characteristic clinical features within 1 to 2 years of
onset. The typical picture of bilateral symmetric inflammatory polyarthritis
involving small and large joints in both the upper and lower extremities with
sparing of the axial skeleton except the cervical spine suggests the diagnosis.
Constitutional features indicative of the inflammatory nature of the disease,
such as morning stiffness, support the diagnosis. Demonstration of subcutaneous
nodules is a helpful diagnostic feature. Additionally, the presence of
rheumatoid factor, inflammatory synovial fluid with increased numbers of
polymorphonuclear leukocytes, and radiographic findings of juxtaarticular bone
demineralization and erosions of the affected joints substantiate the
diagnosis.
The
diagnosis is somewhat more difficult early in the course when only
constitutional symptoms or intermittent arthralgias or arthritis in an
asymmetric distribution may be present. A period of observation may be
necessary before the diagnosis can be established. A definitive diagnosis of RA
depends predominantly on characteristic clinical features and the exclusion of
other inflammatory processes. The isolated finding of a positive test for rheumatoid
factor or an elevated erythrocyte sedimentation rate, especially in an older
person with joint pains, should not itself be used as evidence of RA.
In
1987, the American College of Rheumatology developed revised criteria for the
classification of RA (Table 312-1). These criteria demonstrate a sensitivity of
91 to 94% and a specificity of 89% when used to classify patients with RA
compared with control subjects with rheumatic diseases other than RA. Although
these criteria were developed as a means of disease classification for
epidemiologic purposes, they can be useful as guidelines for establishing the
diagnosis. Failure to meet these criteria, however, especially during the early
stages of the disease, does not exclude the diagnosis. Moreover, in patients
with early arthritis, the criteria do not discriminate effectively between
patients who subsequently develop persistent, disabling, or erosive disease and
those who do not.
Table 312-1: The 1987 Revised Criteria for the
Classification of Rheumatoid Arthritis
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|||
![]() Treatment
Treatment
General Principles
The
goals of therapy of RA are (1) relief of pain, (2) reduction of inflammation,
(3) protection of articular structures, (4) maintenance of function, and (5)
control of systemic involvement. Since the etiology of RA is unknown, the
pathogenesis is not completely delineated, and the mechanisms of action of many
of the therapeutic agents employed are uncertain, therapy remains largely
empirical. None of the therapeutic interventions is curative, and therefore all
must be viewed as palliative, aimed at relieving the signs and symptoms of the
disease. The various therapies employed are directed at nonspecific suppression
of the inflammatory or immunologic process in the hope of ameliorating symptoms
and preventing progressive damage to articular structures.
Management
of patients with RA involves an interdisciplinary approach, which attempts to
deal with the various problems that these individuals encounter with functional
as well as psychosocial interactions. A variety of physical therapy modalities
may be useful in decreasing the symptoms of RA. Rest ameliorates symptoms and
can be an important component of the total therapeutic program. In addition,
splinting to reduce unwanted motion of inflamed joints may be useful. Exercise
directed at maintaining muscle strength and joint mobility without exacerbating
joint inflammation is also an important aspect of the therapeutic regimen. A
variety of orthotic and assistive devices can be helpful in supporting and
aligning deformed joints to reduce pain and improve function. Education of the
patient and family is an important component of the therapeutic plan to help
those involved become aware of the potential impact of the disease and make
appropriate accommodations in life-style to maximize satisfaction and minimize
stress on joints.
Medical
management of RA involves five general approaches. The first is the use of
aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) and simple
analgesics to control the symptoms and signs of the local inflammatory process.
These agents are rapidly effective at mitigating signs and symptoms, but they
appear to exert minimal effect on the progression of the disease. Recently,
specific inhibitors of the isoform of cyclooxygenase (Cox) that is upregulated
at inflammatory sites (Cox-2) have been developed. Cox-2-specific inhibitors
(CSIs) have been shown to be as effective as classic NSAIDs, which inhibit both
isoforms of Cox, but to cause significantly less gastroduodenal ulceration. The
second line of therapy involves use of low-dose oral glucocorticoids. Although
low-dose glucocorticoids have been widely used to suppress signs and symptoms
of inflammation, recent evidence suggests that they may also retard the
development and progression of bone erosions. Intraarticular glucocorticoids
can often provide transient symptomatic relief when systemic medical therapy
has failed to resolve inflammation. The third line of agents includes a variety
of agents that have been classified as the disease-modifying or slow-acting
antirheumatic drugs. These agents appear to have the capacity to decrease
elevated levels of acute-phase reactants in treated patients and, therefore,
are thought to modify the inflammatory component of RA and thus its destructive
capacity. Recently, combinations of disease-modifying antirheumatic drugs
(DMARDs) have shown promise in controlling the signs and symptoms of RA. A
fourth group of agents are the TNF-![]() neutralizing agents, which have been shown to have a major impact on the signs
and symptoms of RA. A fifth group of agents are the immunosuppressive and
cytotoxic drugs that have been shown to ameliorate the disease process in some
patients. Additional approaches have been employed in an attempt to control the
signs and symptoms of RA. Substituting omega-3 fatty acids such as
eicosapentaenoic acid found in certain fish oils for dietary omega-6 essential
fatty acids found in meat has also been shown to provide symptomatic
improvement in patients with RA. A variety of nontraditional approaches have
also been claimed to be effective in treating RA, including diets, plant and
animal extracts, vaccines, hormones, and topical preparations of various sorts.
Many of these are costly, and none has been shown to be effective. However,
belief in their efficacy ensures their continued use by some patients.
neutralizing agents, which have been shown to have a major impact on the signs
and symptoms of RA. A fifth group of agents are the immunosuppressive and
cytotoxic drugs that have been shown to ameliorate the disease process in some
patients. Additional approaches have been employed in an attempt to control the
signs and symptoms of RA. Substituting omega-3 fatty acids such as
eicosapentaenoic acid found in certain fish oils for dietary omega-6 essential
fatty acids found in meat has also been shown to provide symptomatic
improvement in patients with RA. A variety of nontraditional approaches have
also been claimed to be effective in treating RA, including diets, plant and
animal extracts, vaccines, hormones, and topical preparations of various sorts.
Many of these are costly, and none has been shown to be effective. However,
belief in their efficacy ensures their continued use by some patients.
Drugs
Nonsteroidal Anti-Inflammatory Drugs
Besides
aspirin, many NSAIDs are available to treat RA. As a result of the capacity of
these agents to block the activity of the Cox enzymes and therefore the
production of prostaglandins, prostacyclin, and thromboxanes, they have
analgesic, anti-inflammatory, and antipyretic properties. In addition, the
agents may exert other anti-inflammatory effects. These agents are all
associated with a wide spectrum of toxic side effects. Some, such as gastric
irritation, azotemia, platelet dysfunction, and exacerbation of allergic
rhinitis and asthma, are related to the inhibition of cyclooxygenase activity,
whereas a variety of others, such as rash, liver function abnormalities, and
bone marrow depression, may not be. None of the NSAIDs has been shown to be
more effective than aspirin in the treatment of RA. However, these nonaspirin
drugs are associated with a lower incidence of gastrointestinal intolerance.
None of the newer NSAIDs appears to show significant therapeutic advantages
over the other available agents. In addition, there is no consistent advantage
of any of these newer agents over the others with respect to the incidence or
severity of toxic manifestations. Recent evidence indicates that two separate
enzymes, Cox-1 and -2, are responsible for the initial metabolism of
arachidonic acid into various inflammatory mediators. The former is
constitutively present in many cells and tissues, including the stomach and the
platelet, whereas the latter is specifically induced in response to
inflammatory stimuli. Inhibition of Cox-2 accounts for the anti-inflammatory
effects of NSAIDs, whereas inhibition of Cox-1 induces much of the
mechanism-based toxicity. As the currently available NSAIDs inhibit both
enzymes, therapeutic benefit and toxicity are intertwined. CSIs have now been
approved for the treatment of RA. Clinical trials have shown that CSIs suppress
the signs and symptoms of RA as effectively as classic Cox-nonspecific NSAIDs
but are associated with a significantly reduced incidence of gastroduodenal
ulceration. This suggests that CSIs might be considered instead of classic
Cox-nonspecific NSAIDs, especially in persons with increased risk of
NSAID-induced major upper gastrointestinal side effects, including persons over
65, those with a history of peptic ulcer disease, persons receiving
glucocorticoids or anticoagulants, or those requiring high doses of NSAIDs.
Disease-Modifying Antirheumatic Drugs
Clinical
experience has delineated a number of agents that appear to have the capacity
to alter the course of RA. This group of agents includes methotrexate, gold
compounds, D-penicillamine, the antimalarials, and sulfasalazine. Despite
having no chemical or pharmacologic similarities, in practice these agents
share a number of characteristics. They exert minimal direct nonspecific
anti-inflammatory or analgesic effects, and therefore NSAIDs must be continued
during their administration, except in a few cases when true remissions are
induced with them. The appearance of benefit from DMARD therapy is usually
delayed for weeks or months. As many as two-thirds of patients develop some
clinical improvement as a result of therapy with any of these agents, although
the induction of true remissions is unusual. In addition to clinical
improvement, there is frequently an improvement in serologic evidence of
disease activity, and titers of rheumatoid factor and C-reactive protein and
the erythrocyte sedimentation rate frequently decline. Moreover, emerging
evidence suggests that DMARDs actually retard the development of bone erosions
or facilitate their healing. Furthermore, developing evidence suggests that
early aggressive treatment with DMARDs may be effective at slowing the
appearance of bone erosions.
Which
DMARD should be the drug of first choice remains controversial, and trials have
failed to demonstrate a consistent advantage of one over the other. Despite
this, methotrexate has emerged as the DMARD of choice because of its relatively
rapid onset of action, its capacity to effect sustained improvement with
ongoing therapy, and the high level of patient retention on therapy. Each of
the DMARDs is associated with considerable toxicity, and therefore careful
patient monitoring is necessary. Toxicity of the various agents also becomes
important in determining the drug of first choice. Of note, failure to respond
or development of toxicity to one DMARD does not preclude responsiveness to
another. Thus, a similar percentage of RA patients who have failed to respond
to one DMARD will respond to another when it is given as the second
disease-modifying drug.
No
characteristic features of patients have emerged that predict responsiveness to
a DMARD. Moreover, the indications for the initiation of therapy with one of
these agents are not well defined, although recently the trend has been to
begin DMARD therapy early in the course of the disease, and data have begun to
emerge to support the conclusion that this approach may slow the development of
bone erosions, although this remains controversial.
The
folic acid antagonist methotrexate, given in an intermittent low dose (7.5 to 30
mg once weekly), is currently a frequently utilized DMARD. Most rheumatologists
recommend use of methotrexate as the initial DMARD, especially in individuals
with evidence of aggressive RA. Recent trials have documented the efficacy of
methotrexate and have indicated that its onset of action is more rapid than
other DMARDs, and patients tend to remain on therapy with methotrexate longer
than they remain on other DMARDs because of better clinical responses and less
toxicity. Long-term trials have indicated that methotrexate does not induce
remission but rather suppresses symptoms while it is being administered.
Maximal improvement is observed after 6 months of therapy, with little
additional improvement thereafter. Major toxicity includes gastrointestinal upset,
oral ulceration, and liver function abnormalities that appear to be
dose-related and reversible and hepatic fibrosis that can be quite insidious,
requiring liver biopsy for detection in its early stages. Drug-induced
pneumonitis has also been reported. Liver biopsy is recommended for individuals
with persistent or repetitive liver function abnormalities. Concurrent
administration of folic acid or folinic acid may diminish the frequency of some
side effects without diminishing effectiveness.
Glucocorticoid Therapy
Systemic
glucocorticoid therapy can provide effective symptomatic therapy in patients
with RA. Low-dose (<7.5 mg/d) prednisone has been advocated as useful
additive therapy to control symptoms. Moreover, recent evidence suggests that
low-dose glucocorticoid therapy may retard the progression of bone erosions.
Monthly pulses with high-dose glucocorticoids may be useful in some patients
and may hasten the response when therapy with a DMARD is initiated.
TNF-![]() neutralizing agents
neutralizing agents
Recently,
biologic agents that bind and neutralize TNF-![]() have become available. One of these is a TNF-
have become available. One of these is a TNF-![]() type II receptor fused to IgG1 (etanercept), and the second is a chimeric
mouse/human monoclonal antibody to TNF-
type II receptor fused to IgG1 (etanercept), and the second is a chimeric
mouse/human monoclonal antibody to TNF-![]() (infliximab). Clinical trials have shown that parenteral administration of
either TNF-
(infliximab). Clinical trials have shown that parenteral administration of
either TNF-![]() neutralizing agent is remarkably effective at controlling signs and symptoms of
RA in patients who have failed DMARD therapy. Repetitive therapy with these
agents is effective with or without concomitant methotrexate. Although these
agents are notably effective in persistently controlling signs and symptoms of
RA in a majority of patients, their impact on the progression of bone erosions
has not been proven. Side effects include the potential for an increased risk
of serious infections and the development of anti-DNA antibodies, but with no
associated evidence of signs and symptoms of systemic lupus erythematosus.
Although these side effects are uncommon, their occurrence mandates that TNF-
neutralizing agent is remarkably effective at controlling signs and symptoms of
RA in patients who have failed DMARD therapy. Repetitive therapy with these
agents is effective with or without concomitant methotrexate. Although these
agents are notably effective in persistently controlling signs and symptoms of
RA in a majority of patients, their impact on the progression of bone erosions
has not been proven. Side effects include the potential for an increased risk
of serious infections and the development of anti-DNA antibodies, but with no
associated evidence of signs and symptoms of systemic lupus erythematosus.
Although these side effects are uncommon, their occurrence mandates that TNF-![]() neutralizing therapy be supervised by physicians with experience in their use.
neutralizing therapy be supervised by physicians with experience in their use.
Immunosuppressive Therapy
The
immunosuppressive drugs azathioprine, leflunomide, cyclosporine, and
cyclophosphamide have been shown to be effective in the treatment of RA and to
exert therapeutic effects similar to those of the DMARDs. However, these agents
appear to be no more effective than the DMARDs. Moreover, they cause a variety
of toxic side effects, and cyclophosphamide appears to predispose the patient
to the development of malignant neoplasms. Therefore, these drugs have been
reserved for patients who have clearly failed therapy with DMARDs. On occasion,
extraarticular disease such as rheumatoid vasculitis may require cytotoxic
immunosuppressive therapy.
Surgery
Surgery
plays a role in the management of patients with severely damaged joints.
Although arthroplasties and total joint replacements can be done on a number of
joints, the most successful procedures are carried out on hips, knees, and shoulders.
Realistic goals of these procedures are relief of pain and reduction of
disability. Reconstructive hand surgery may lead to cosmetic improvement and
some functional benefit. Open or arthroscopic synovectomy may be useful in some
patients with persistent monarthritis, especially of the knee. Although
synovectomy may offer short-term relief of symptoms, it does not appear to
retard bone destruction or alter the natural history of the disease. In
addition, early tenosynovectomy of the wrist may prevent tendon rupture.
![]() Approach to
the Patient
Approach to
the Patient
An
approach to the medical management of patients with RA is depicted in Fig.
312-3. The principles underlying care of these patients reflect the variability
of the disease, the frequent persistent nature of the inflammation and its
potential to cause disability, the relationship between sustained inflammation
and bone erosions, and the need to reevaluate the patient frequently for
symptomatic response to therapy, progression of disability and joint damage,
and side effects of treatment. At the onset of disease it is difficult to
predict the natural history of an individual patient's illness. Therefore, the
usual approach is to attempt to alleviate the patient's symptoms with NSAIDs or
CSIs. Some patients may have mild disease that requires no additional therapy.
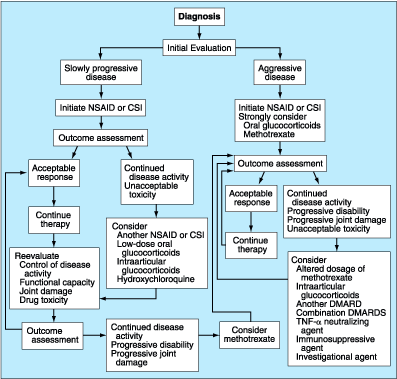
Figure 312-3: Algorithm for the medical management of
rheumatoid arthritis. NSAID, nonsteroidal anti-inflammatory drug; CSI, Cox-2
specific inhibitor; DMARD, disease-modifying antirheumatic drug; TNF-![]() ,
tumor necrosis factor
,
tumor necrosis factor ![]() .
.
At
some time during most patients' course, the possibility of initiating DMARD
therapy and/or low-dose oral glucocorticoids is entertained. With aggressive
disease this might occur sooner, often within 1 to 3 months of diagnosis,
whereas in patients with more indolent disease, smoldering activity may not
require such therapy for many years. The development of bone erosions or
radiographic evidence of cartilage loss is clear-cut evidence of the
destructive potential of the inflammatory process and indicates the need for
DMARD therapy. The other indications as outlined above, including persistent
pain, joint swelling, or functional impairment, are much more subjective,
however. As persistent inflammation, involvement of multiple joints, elevated
levels of acute-phase reactants, and rheumatoid factor titers correlate with
the development of disability and/or bony erosions, some have advocated the use
of these prognostic indicators of aggressive disease in the decision to employ DMARDs
early in the course of RA. The decision to begin use of a DMARD and/or low-dose
oral glucocorticoids requires experience and clinical judgment as well as the
ability to assess joint swelling and functional activity and the patient's pain
tolerance and expectation of therapy accurately. In this setting, the fully
informed patient must play an active role in the decision to begin DMARD and/or
low-dose oral glucocorticoid therapy, after careful review of the therapeutic
and toxic potential of the various drugs.
If
a patient responds to a DMARD, therapy is continued with careful monitoring to
avoid toxicity. All DMARDs provide a suppressive effect and therefore require
prolonged administration. Even with successful therapy, local injection of
glucocorticoids may be necessary to diminish inflammation that may persist in a
limited number of joints. In addition, NSAIDs or CSIs may be necessary to
mitigate symptoms. Even after inflammation has totally resolved, symptoms from
loss of cartilage and supervening degenerative joint disease or altered joint
function may require additional treatment. Surgery may also be necessary to
relieve pain or diminish the functional impairment secondary to alterations in
joint function. Recently an alternative approach to treat patients with RA has
been suggested. This involves the initiation of therapy with multiple agents
early in the course of disease in an attempt to control inflammation, followed
by maintenance on one or more agents as necessary to control disease activity. The
effectiveness of this therapeutic alternative has not been proved.