The Bradyarrhythmias: Disorders of Sinus Node Function
and AV Conduction Disturbances
Anatomy of the Conducting System
Under
normal conditions, the pacemaker function of the heart resides in the
sinoatrial (SA) node, which lies at the junction of the right atrium and
superior vena cava. The SA node is approximately 1.5 cm long and 2 to 3 mm wide
and is supplied by the sinus node artery, which arises from either the right
coronary artery (60%) or the left circumflex coronary artery (40%). Once the
impulse exits the sinus node and perinodal tissue, it traverses the atrium
until it reaches the atrioventricular (AV) node. The blood supply of the AV
node is derived from the posterior descending coronary artery (90%). The AV
node lies at the base of the interatrial septum just above the tricuspid
annulus and anterior to the coronary sinus. The electrophysiologic properties
of the AV node result in slow conduction, which is responsible for the normal
delay in AV conduction, i.e., the PR interval.
The
bundle of His emerges from the AV node, enters the fibrous skeleton of the
heart, and courses anteriorly across the membranous interventricular septum. It
has a dual blood supply from the AV nodal artery and a branch of the anterior
descending coronary artery. The branching (distal) portion of the bundle of His
gives rise to a broad sheet of fibers that course over the left side of the
interventricular septum to form the left bundle branch and a narrow cable-like
structure on the right side that forms the right bundle branch. The
arborization of both the right and left bundle branches gives rise to the
distal His-Purkinje system, which ultimately extends throughout the endocardium
of the right and left ventricles.
The
SA node, atrium, and AV node are significantly influenced by autonomic tone.
Vagal influences depress automaticity of the SA node, depress conduction, and
prolong refractoriness in the tissue surrounding the SA node; inhomogeneously
decrease atrial refractoriness and slow atrial conduction; and prolong AV nodal
conduction and refractoriness. Sympathetic influences exert the opposite
effect.
Electrophysiologic Principles
In
the resting state, the interior of most cardiac cells, with the exception of
the SA and AV nodes, is approximately -80 to -90 mV, negative with respect to a
reference extracellular electrode. The resting membrane potential is determined
primarily by the concentration gradient of potassium across the cell membrane.
Activation of cardiac cells results from movement of ions across the cell
membrane, causing a transient depolarization known as the action potential.
The ionic species responsible for the action potential varies among the cardiac
tissues, and the configuration of the action potential is therefore unique to
each tissue (Fig. 229-1).
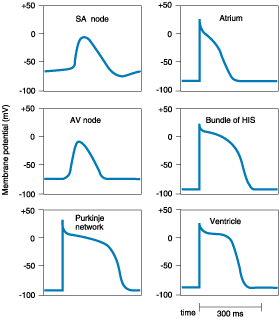
Figure 229-1: Action potential configurations in
different regions of the mammalian heart.
The
action potential of the His-Purkinje system and ventricular myocardium has five
phases (Fig. 229-2). The rapid depolarizing current (phase 0) is mainly
determined by an influx of sodium into myocardial cells followed by a secondary
(slower) influx of calcium, which produces a slow inward current. The
repolarization phases of the action potential (phases 1 to 3) are primarily
related to outward flux of potassium. The resting membrane potential is phase
4.
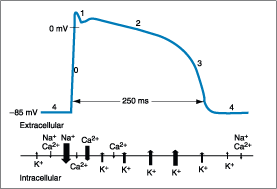
Figure 229-2: Schematic representation of the action
potential in normal ventricle depicting the direction, strength, and period of
flow of the ionic currents underlying the action potential. The arrow's
direction and size indicate whether current is inward- or outward-directed and
the approximate current strength of the ion identified at the arrow's base. The
horizontal position of the arrow corresponds to the same moment in the time
course of action potential (see text). The five phases of the action potential
are indicated by the numerals placed along the waveform.
The
bradyarrhythmias result from abnormalities either of impulse formation, i.e.,
automaticity, or of conduction. Automaticity, which is normally observed
in the sinus node, the specialized fibers of the His-Purkinje system, and some
specialized atrial fibers, is the property of a cardiac cell that causes it to
depolarize spontaneously during phase 4 of the action potential, leading to the
generation of an impulse. To exhibit automaticity, the resting membrane
potential must decrease spontaneously until threshold potential is reached and
an all-or-none regenerative response occurs. The ionic currents producing
spontaneous diastolic depolarization appear to involve the inward current of
either sodium or calcium and a decreasing outward potassium current. The
velocity of conduction, i.e., impulse propagation through cardiac
tissues, depends on the magnitude of inward current, which is directly related
to the rate of rise and amplitude of phase 0 of the action potential. The more
positive the threshold potential and the slower the rate of depolarization
toward threshold, the slower is the rate of rise of phase 0 of the action
potential and the slower is the conduction velocity. Disease states or drugs
may result in lower rates of rise of phase 0 at any given membrane potential.
Passive membrane properties (e.g., intracellular resistance and intercellular
coupling) can also affect impulse propagation. Propagation is more rapid
parallel to fiber orientation than transverse to it, a property termed anisotropic
conduction.
Refractoriness is a property of cardiac cells that defines the
period of recovery that cells require after being discharged before they can be
reexcited by a stimulus. The absolute refractory period is defined by
that portion of the action potential during which no stimulus, regardless of
its strength, can evoke another response. The effective refractory period
is that part of the action potential during which a stimulus can evoke only a
local, nonpropagated response. The relative refractory period extends
from the end of the effective refractory period to the time that the tissue is
fully recovered. During this time, a stimulus of greater than threshold
strength is required to evoke a response, which is propagated more slowly than
normal. In the normal His-Purkinje system or ventricular myocytes, excitability
is recovered following completion of the action potential, and evoked responses
have characteristics similar to the spontaneous normal response. In the AV
node, recovery of excitability occurs well after completion of the action
potential.
Intracardiac Recordings of the Specialized
Conducting System
Electrode
catheters allow the recording of activation of portions of the specialized
conducting system, including the bundle of His. To obtain a recording from the
bundle of His, the electrode catheter is positioned across the tricuspid valve
(Fig. 229-3). The interval from local atrial depolarization in the His bundle
recording to the onset of depolarization of the His bundle deflection is called
the AH interval (normal = 60 to 125 ms) and represents an indirect
method of assessing AV nodal conduction time. The interval from the beginning
of the His bundle deflection to the earliest onset of ventricular activation,
as measured from any of multiple-surface electrocardiogram (ECG) leads or the
intracardiac ventricular electrogram, is called the HV interval (normal
= 35 to 55 ms) and represents conduction time through the His-Purkinje system.
Electrode catheters can be positioned in the area of the sinus node to record
high right atrial activity. Left atrial activity may be recorded directly via a
catheter placed across a patent foramen ovale or indirectly using a catheter
inserted into the coronary sinus. The atrial activation sequence may be
"mapped," and sites of intra- and interatrial conduction
abnormalities may be ascertained.
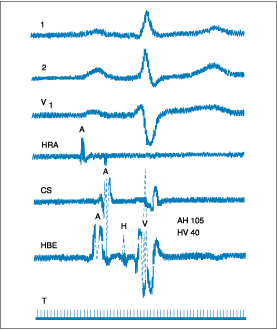
Figure 229-3:
Sinus Node Dysfunction
The
SA node is normally the dominant cardiac pacemaker because its intrinsic
discharge rate is the highest of all potential cardiac pacemakers. Its
responsiveness to alterations in autonomic nervous system tone is responsible
for the normal acceleration of heart rate during exercise and the slowing that
occurs during rest and sleep. Increases in sinus rate normally result from an
increase in sympathetic tone acting via ![]() -adrenergic
receptors and/or a decrease in parasympathetic tone acting via muscarinic
receptors. Slowing of the heart rate is normally due to opposite alterations.
In adults, the normal sinus rate under basal conditions is 60 to 100 beats per
minute. Sinus bradycardia is said to exist when the sinus rate is less
than 60 beats per minute, and sinus tachycardia when it exceeds 100
beats per minute. However, there is wide variation among individuals, and rates
less than 60 beats per minute do not necessarily indicate pathologic states.
For example, trained athletes often exhibit resting rates under 50 beats per
minute due to increases in vagal tone. Normal elderly individuals may also show
marked sinus bradycardia at rest.
-adrenergic
receptors and/or a decrease in parasympathetic tone acting via muscarinic
receptors. Slowing of the heart rate is normally due to opposite alterations.
In adults, the normal sinus rate under basal conditions is 60 to 100 beats per
minute. Sinus bradycardia is said to exist when the sinus rate is less
than 60 beats per minute, and sinus tachycardia when it exceeds 100
beats per minute. However, there is wide variation among individuals, and rates
less than 60 beats per minute do not necessarily indicate pathologic states.
For example, trained athletes often exhibit resting rates under 50 beats per
minute due to increases in vagal tone. Normal elderly individuals may also show
marked sinus bradycardia at rest.
Etiology
SA
node dysfunction is most often found in the elderly as an isolated phenomenon.
Although interruption of the blood supply to the SA node may produce
dysfunction, the correlation between obstruction of the sinus node artery and
clinical evidence of SA node dysfunction is poor. Specific disease states
associated with SA node dysfunction include senile amyloidosis and other
conditions associated with infiltration of the atrial myocardium. Sinus
bradycardia is associated with hypothyroidism, advanced liver disease,
hypothermia, typhoid fever, and brucellosis; it occurs during episodes of
hypervagotonia (vasovagal syncope), severe hypoxia, hypercapnia, acidemia, and
acute hypertension. However, most cases of SA node dysfunction are due to
idiopathic degeneration or are secondary to pharmacologic agents.
Manifestations
Although
marked (![]() 50
beats per minute) sinus bradycardia may cause fatigue and other symptoms due to
inadequate cardiac output, more commonly sinus node dysfunction is manifest as
paroxysmal dizziness, presyncope, or syncope. These symptoms usually result
from abrupt, prolonged sinus pauses caused by failure of sinus impulse
formation (sinus arrest) or block of conduction of sinus impulses to the
surrounding atrial tissue (sinus exit block). In either case, the ECG
manifestation is a prolonged period (>3 s) of atrial asystole. In some
patients, SA node dysfunction is accompanied by abnormalities in AV conduction.
In addition to the absence of atrial activity, lower pacemakers fail to emerge
during the sinus pauses, resulting in periods of ventricular asystole and
syncope. Occasionally, SA node dysfunction is manifested by an inadequate
acceleration in sinus rate in response to a stress such as exercise or fever.
In some patients, SA node dysfunction may become manifest only in the presence
of certain cardioactive drugs: cardiac glycosides,
50
beats per minute) sinus bradycardia may cause fatigue and other symptoms due to
inadequate cardiac output, more commonly sinus node dysfunction is manifest as
paroxysmal dizziness, presyncope, or syncope. These symptoms usually result
from abrupt, prolonged sinus pauses caused by failure of sinus impulse
formation (sinus arrest) or block of conduction of sinus impulses to the
surrounding atrial tissue (sinus exit block). In either case, the ECG
manifestation is a prolonged period (>3 s) of atrial asystole. In some
patients, SA node dysfunction is accompanied by abnormalities in AV conduction.
In addition to the absence of atrial activity, lower pacemakers fail to emerge
during the sinus pauses, resulting in periods of ventricular asystole and
syncope. Occasionally, SA node dysfunction is manifested by an inadequate
acceleration in sinus rate in response to a stress such as exercise or fever.
In some patients, SA node dysfunction may become manifest only in the presence
of certain cardioactive drugs: cardiac glycosides, ![]() -adrenergic
blocking drugs, calcium channel blockers, amiodarone, and other antiarrhythmic
agents. These agents, which do not usually cause sinus node dysfunction in
normal people, may unmask evidence of sinus node dysfunction in susceptible
individuals.
-adrenergic
blocking drugs, calcium channel blockers, amiodarone, and other antiarrhythmic
agents. These agents, which do not usually cause sinus node dysfunction in
normal people, may unmask evidence of sinus node dysfunction in susceptible
individuals.
The
sick sinus syndrome refers to a combination of symptoms (dizziness,
confusion, fatigue, syncope, and congestive heart failure) caused by SA node
dysfunction and manifested by marked sinus bradycardia, sinoatrial block, or
sinus arrest. Because these symptoms are nonspecific, and because ECG
manifestations of sinus node dysfunction are often intermittent, it may be
difficult to prove that such symptoms are actually caused by SA node
dysfunction.
Atrial
tachyarrhythmias such as atrial fibrillation, atrial flutter, or atrial tachycardia
may be accompanied by SA node dysfunction. The bradycardia-tachycardia
syndrome refers to paroxysmal atrial arrhythmia that upon termination is
followed by prolonged sinus pauses (Fig. 229-4) or in which there are
alternating periods of tachyarrhythmia and bradyarrhythmia. Syncope or
presyncope may result from failure of the sinus node to recover function
following suppression of automaticity by atrial tachyarrhythmia.
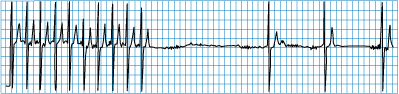
Figure 229-4: Tachycardia-bradycardia syndrome. Rhythm
strip of ECG lead II showing spontaneous cessation of supraventricular
tachycardia followed by a 6-s pause prior to resumption of sinus activity. The
patient was asymptomatic during supraventricular tachycardia, but the sinus
pause caused severe light-headedness.
Diagnosis
First-degree sinoatrial exit block denotes a prolonged
conduction time from the SA node to the surrounding atrial tissue. It cannot be
recognized on a standard (surface) ECG but requires invasive intracardiac
recordings, which can detect this condition indirectly, by measuring the sinus
response to atrial premature beats, or directly, by recording SA node
electrograms. Second-degree sinoatrial exit block denotes the
intermittent failure of conduction of sinus impulses to the surrounding atrial
tissue; it is manifested as the intermittent absence of P waves (Fig. 229-5). Third-degree,
or complete, sinoatrial block is characterized by a lack of atrial
activity or by the presence of an ectopic subsidiary atrial pacemaker. On the
standard ECG it cannot be distinguished from sinus arrest, but direct
intracardiac recordings of SA node activity permit this distinction. The bradycardia-tachycardia
syndrome is manifested on the standard ECG as tachyarrhythmias (Fig.
229-4). Most often these are atrial flutter or fibrillation, although any
tachycardia during which the atria are activated may cause overdrive
suppression of the sinus node resulting in clinical appearance of this
syndrome.

Figure 229-5: Second-degree sinoatrial exit block.
Surface ECG denoting abrupt absence of P wave during sinus rhythm. Prior to the
pause, the sinus rate is regular. The interval of the pause is exactly twice
the basal sinus cycle length. The arrow marks the appropriate location for the
absent P wave. SA exit block can be 2:1 as above or longer, as shown in Fig.
229-6.
The
most important step in the diagnosis is to correlate symptoms with ECG evidence
of SA node dysfunction. While ambulatory ECG (Holter) monitoring remains a
mainstay in evaluating sinus node function, most episodes of syncope are paroxysmal
and unpredictable. Single and even multiple 24-h Holter monitor recordings may
fail to include a symptomatic episode.
Caution
must be taken in interpreting the Holter monitor results. For instance, a pause
during sleep is often a normal finding associated with heightened vagal tone.
This should not be interpreted as sinus node dysfunction requiring pacemaker
implantation.
Continuous-loop
event records represent a more specific diagnostic tool. These devices may be
worn for prolonged periods of time and allow close correlation between
electrocardiographic findings and symptoms. They do require the patient's
ability to activate the monitor at the time of symptoms. More recently, an
implantable event recorder, which can be interrogated like a pacemaker, has
been developed for patients with rare events.
The
response to carotid sinus pressure and pharmacologic autonomic
"denervation" of the heart may be helpful. Carotid sinus pressure can
be particularly useful in patients in whom paroxysmal dizziness or syncope is
compatible with the hypersensitive carotid sinus syndrome (Chap. 21). In such
patients, the response can be dramatic, and sinus pauses in excess of 5 s may
occur. Although pauses in excess of 3 s are considered abnormal, in elderly
patients such pauses are common and do not necessarily signify a diagnostic
response. This is a major limitation of the use of carotid sinus pressure as a
diagnostic test in the elderly. The other noninvasive test of SA node function
involves the use of pharmacologic agents to manipulate the autonomic nervous
system and assess the balance of parasympathetic and sympathetic activity on
the sinus node. Physiologic or pharmacologic maneuvers that are vagomimetic
(Valsalva maneuver or phenylephrine-induced hypertension), vagolytic
(atropine), sympathomimetic (isoproterenol or hypotension by nitroprusside), or
sympatholytic (![]() -adrenergic
blocking agents) can be utilized, singly and in combination. These studies are
designed to test the response of the sinus node to autonomic stimulation and
inhibition and thereby characterize the status of autonomic regulation of the
sinus node. Abnormalities of the autonomic control of sinus function are
particularly common in patients in whom asymptomatic sinus bradycardia is
documented.
-adrenergic
blocking agents) can be utilized, singly and in combination. These studies are
designed to test the response of the sinus node to autonomic stimulation and
inhibition and thereby characterize the status of autonomic regulation of the
sinus node. Abnormalities of the autonomic control of sinus function are
particularly common in patients in whom asymptomatic sinus bradycardia is
documented.
Intrinsic Heart Rate
This
is a manifestation of the primary activity of the SA node, and its
determination requires chemical autonomic blockade of the heart with a
combination of atropine and a beta blocker. Normal values of intrinsic heart
rate (in beats per minute) are calculated by the formula 118.1 - (0.57 × age).
The use of autonomic blockade can separate patients with asymptomatic sinus
bradycardia into a group with primary sinus node dysfunction (slow intrinsic
heart rate) and a group with autonomic imbalance (normal intrinsic heart rate).
Autonomic blockade is particularly useful when combined with invasive
assessment of sinus node function. Autonomic blockade may depress conduction in
patients with intrinsic disease of the conduction system and should be carried
out only in a setting where arrhythmias can be monitored and treated rapidly.
Evaluation
The
invasive electrophysiologic investigation of SA node dysfunction should be
undertaken in patients who have had symptoms compatible with SA node
dysfunction and in whom no documentation of the arrhythmia responsible for
these symptoms has been obtained by prolonged Holter monitoring. Asymptomatic
patients with sinus bradycardia need not be tested, since no therapy is
indicated. Similarly, symptomatic patients with ECG documentation of asystole,
sinoatrial block or arrest, or the bradycardia-tachycardia syndrome do not
require electrophysiologic tests for diagnosis. However, in symptomatic
patients without documentation of an arrhythmia, electrophysiologic assessment
of SA node function can yield information that may be used to guide appropriate
therapy.
The
results of electrophysiologic tests of sinus node function must be interpreted
with caution. SA node dysfunction coexists frequently with other disorders such
as AV conduction disturbances, which may cause symptoms such as syncope.
Electrophysiologic evaluation of patients with symptoms such as undiagnosed
syncope must not stop with the demonstration of abnormalities of SA node
dysfunction or carotid sinus hypersensitivity. Instead, complete evaluation,
including His bundle recordings and programmed atrial and ventricular stimulation
(Chap. 230), is necessary to search for additional electrophysiologic
abnormalities that could be responsible for symptoms.
Treatment
Permanent
pacemakers (see Table 229-4) are the mainstay of therapy for patients with
symptomatic SA node dysfunction. Patients with intermittent paroxysms of
bradycardia or sinus arrest and with the cardioinhibitory form of the
hypersensitive carotid sinus syndrome are usually adequately treated by demand
ventricular pacemakers. These devices are reliable, relatively inexpensive, and
suffice to prevent episodic symptoms due to abrupt bradycardia. Whether
dual-chamber pacing offers any advantages to ventricular pacing in such
circumstances remains uncertain. Patients with symptomatic chronic sinus
bradycardia or frequent prolonged episodes of sinus node dysfunction do better
with dual-chamber pacemakers that preserve the normal AV activation sequence.
Although theoretically an atrial demand pacemaker should be adequate for
patients with SA node dysfunction, the frequent accompaniment of dysfunction in
other portions of the cardiac conduction system usually mandates placement of a
pacemaker capable of ventricular pacing. Recent studies suggest that AV
sequential pacing may also be useful in preventing atrial fibrillation, an
important component of the bradycardia-tachycardia syndrome.
Table 229-4: Guidelines for Permanent Pacing*
|
|||||||||||||||||||||||||||||||||||
|
AV
Conduction Disturbances
The
specialized cardiac conducting system normally ensures synchronous conduction
of each sinus impulse from the atria to the ventricles. Abnormalities of
conduction of the sinus impulse to the ventricles may portend the development
of heart block, which can ultimately lead to syncope or cardiac arrest. In
order to evaluate the clinical significance of conduction abnormalities, the
physician must assess (1) the site of conduction disturbance, (2) the risk of
progression to complete block, and (3) the probability that a subsidiary escape
rhythm arising distal to the site of block will be electrophysiologically and
hemodynamically stable. This latter point is perhaps the most important, since
the rate and stability of the escape pacemaker determine what symptoms result
from heart block. The escape pacemaker following AV nodal block is usually in
the His bundle, which generally has a stable rate of 40 to 60 beats per minute
and is associated with a QRS complex of normal duration (in the absence of a
preexisting intraventricular conduction defect). This contrasts with escape
rhythms arising in the distal His-Purkinje system, which have lower intrinsic
rates (25 to 45 beats per minute), manifest wide QRS complexes with prolonged
duration, and are unstable. Thus, the most important issue is to assess the
risk of infra- or intra-His block (which always mandates a pacemaker) or AV
nodal block in which the frequency of the escape pacemaker is not sufficient to
meet hemodynamic requirements (Table 229-1). Although prolonged QRS complexes
are invariable when the distal His-Purkinje pacemakers form the escape
mechanism, wide QRS complexes can also coexist with AV nodal block and a His
bundle rhythm. Therefore, QRS morphology alone may not be adequate to identify
the site of block.
Table 229-1: Atrioventricular Conduction
Evaluation
|
Etiology
The
AV node is supplied by the parasympathetic and sympathetic nervous systems and
is sensitive to variations in autonomic tone. Chronic slowing of AV nodal
conduction may be seen in highly trained athletes who have hypervagotonia at
rest. A variety of diseases and drugs can also influence AV nodal conduction.
These include acute processes such as myocardial infarction (particularly
inferior), coronary spasm (usually of the right coronary artery), digitalis
intoxication, excesses of beta and/or calcium blockers, acute infections such
as viral myocarditis, acute rheumatic fever, infectious mononucleosis, and
miscellaneous disorders such as Lyme disease, sarcoidosis, amyloidosis, and
neoplasms, particularly cardiac mesotheliomas. AV nodal block may also be
congenital.
Two
degenerative diseases are commonly responsible for damage to the specialized
conducting system and produce AV block usually associated with bundle branch
block (Chap. 226). In Lev's disease, there is calcification and
sclerosis of the fibrous cardiac skeleton, which frequently involves the aortic
and mitral valves, the central fibrous body, and the summit of the ventricular
septum. Lenegre's disease appears to be a primary sclerodegenerative
disease within the conducting system itself with no involvement of the
myocardium or the fibrous skeleton of the heart. These two diseases are
probably the most common causes of isolated chronic heart block in adults.
Hypertension and aortic and/or mitral stenosis are specific disorders that
either accelerate the degeneration of the conducting system or have a direct
effect by calcification and fibrosis involving the conducting system.
First-degree AV block, more properly termed prolonged AV
conduction, is classically characterized by a PR interval >0.20 s, but
use of this value may be misleading in terms of clinical significance. Since
the PR interval is determined by atrial, AV nodal, and His-Purkinje activation,
delay in any one or more of these structures can contribute to a prolonged PR
interval. In the presence of a QRS complex of normal duration, a PR interval
>0.24 s almost invariably is due to a delay within the AV node. If the QRS
is prolonged, delays may be present at any of the levels mentioned above. Delay
within the His-Purkinje system is always accompanied by a prolonged QRS
duration but can occur with a relatively normal PR interval (Fig. 229-6).
However, as indicated below, it is only with intracardiac recordings that the
exact site of delay can be determined.
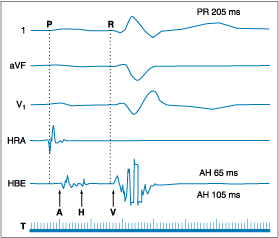
Figure 229-6: Example of marked His-Purkinje system
disease with a relatively normal PR interval. Surface leads I, aVF, and V1
are shown with electrograms from the high right atrium (HRA), His bundle electrogram
(HBE), and time lines (T). The QRS shows right bundle branch block and left
anterior hemiblock; the PR interval is minimally elevated at 205 ms, but the HV
interval exceeds 100 ms. Such a prolonged HV interval mandates a pacemaker.
Second-degree heart block (intermittent AV block) is present when
some atrial impulses fail to conduct to the ventricles. Mobitz type I
second-degree AV block (AV Wenckebach block) is characterized by progressive PR
interval prolongation prior to block of an atrial impulse (Fig. 229-7A).
The pause that follows is less than fully compensatory (i.e., is less than two
normal sinus intervals), and the PR interval of the first conducted impulse is
shorter than the last conducted atrial impulse prior to the blocked P wave. Usually
the difference between the longest and shortest PR intervals exceeds 100 ms.
This type of block is almost always localized to the AV node and associated
with a normal QRS duration, although bundle branch block may be present. It is
seen most often as a transient abnormality with inferior wall infarction or
with drug intoxication, particularly digitalis, beta blockers, and occasionally
calcium channel antagonists. This type of block can also be observed in normal
individuals with heightened vagal tone. Although Mobitz type I block can
progress to complete heart block, this is uncommon, except in the setting of
acute inferior wall myocardial infarction. Even when it does, however, the
heart block is usually well tolerated because the escape pacemaker usually
arises in the proximal His bundle and provides a stable rhythm. As a result,
the presence of Mobitz type I second-degree AV block rarely mandates aggressive
therapy. Therapeutic decisions depend on the ventricular response and the
symptoms of the patient. If the ventricular rate is adequate and the patient is
asymptomatic, observation is sufficient.
|
|
|
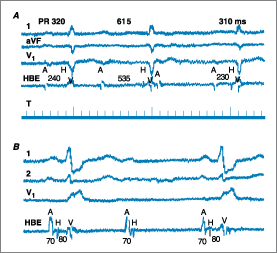
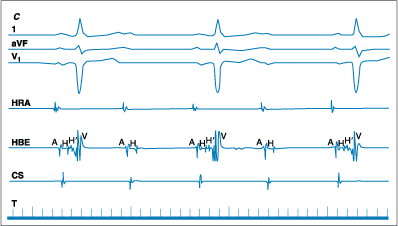
Figure 229-7: A. Mobitz type I
second-degree AV block. Intracardiac recordings demonstrate that the PR
prolongation (320, 615 ms) is localized to the AV node (AH 240, 535 ms,
respectively). HBE, His bundle electrogram; A, atrium; H, His; V, ventricle.
Time lines (T) = 100 ms. B. Mobitz type II second-degree AV block.
Intracardiac recordings document block below the His bundle.
C. 2:1 heart block. Surface leads 1, aVF, V1, demonstrate 2:1
heart block. The intracardiac leads, high right atrium (HRA), coronary sinus
(CS), and HBE, demonstrate a diseased His bundle with marked intra-His
conduction delay (split-His = HH′) during conducted beats and block in
the His bundle every other beat (i.e., between H and H′).
In
Mobitz type II second-degree AV block, conduction fails suddenly and
unexpectedly without a preceding change in PR intervals (Fig. 229-7B).
It is generally due to disease of the His-Purkinje system and is most often
associated with a prolonged QRS duration. When Mobitz type II block occurs with
a normal QRS duration, an intra-His site of block should be expected (Fig.
229-7C). It is important to recognize this type of block because it has
a high incidence of progression to complete heart block with an unstable, slow,
lower escape pacemaker. Therefore, pacemaker implantation is necessary in this
condition. Mobitz type II block may occur in the setting of anteroseptal
infarction or in the primary or secondary sclerodegenerative or calcific
disorders of the fibrous skeleton of the heart. In so-called high-degree AV
block there are periods of two or more consecutively blocked P waves, but
intermittent conduction can be demonstrated. Block is usually in the
His-Purkinje system, but simultaneous block in the AV node may also be present.
Regardless of the site of origin of the escape rhythm, if it is slow and the
patient is symptomatic, a cardiac pacemaker is mandatory.
Third-degree AV block is present when no atrial impulse propagates to
the ventricles. If the QRS complex of the escape rhythm is of normal duration,
occurs at a rate of 40 to 55 beats per minute, and increases with atropine or
exercise, AV nodal block is probable. Congenital complete AV block is usually
localized to the AV node. If the block is within the His bundle, the escape
pacemaker is usually less responsive to these perturbations. If the escape
rhythm of the QRS is wide and associated with rates ![]() 40
beats per minute, block is usually localized in, or distal to, the His bundle
and mandates a pacemaker, since the escape rhythm in this setting is unreliable
(Fig. 229-8). Some patients with infra-His bundle block are capable of
retrograde conduction. In such patients, a "pacemaker syndrome" (see
below) may develop if a simple ventricular pacemaker is used. Dual-chamber
pacemakers eliminate this potential problem.
40
beats per minute, block is usually localized in, or distal to, the His bundle
and mandates a pacemaker, since the escape rhythm in this setting is unreliable
(Fig. 229-8). Some patients with infra-His bundle block are capable of
retrograde conduction. In such patients, a "pacemaker syndrome" (see
below) may develop if a simple ventricular pacemaker is used. Dual-chamber
pacemakers eliminate this potential problem.
AV
Dissociation
AV
dissociation exists whenever the atria and ventricles are under the control of
two separate pacemakers and, while present in complete AV block, can occur in
the absence of a primary conduction disturbance. AV dissociation unrelated to
heart block may occur under two circumstances: First, it may develop with an AV
junctional rhythm in response to severe sinus bradycardia. When the sinus rate
and the escape rate are similar and the P waves occur just before, in, or
following the QRS complex, isorhythmic AV dissociation is said to be
present. Treatment usually consists of removal of the offending cause of sinus
bradycardia (i.e., discontinuation of digitalis, beta blockers, or calcium
antagonists), accelerating the sinus node by vagolytic agents, or insertion of
a pacemaker if the escape rhythm is slow and results in symptoms. Second, AV
dissociation can be caused by an enhanced lower (junctional or ventricular)
pacemaker that competes with normal sinus rhythm and frequently exceeds it.
This has been called interference AV dissociation because the rapid
lower pacemaker results in bombardment of the AV node in a retrograde fashion,
rendering it refractory to the normal sinus impulses. Thus failure of antegrade
conduction is a physiologic response in this circumstance. Interference
dissociation commonly occurs during ventricular tachycardia, accelerated
junctional or ventricular rhythms seen with digitalis intoxication, myocardial
ischemia and/or infarction, or local irritation following cardiac surgery. The
accelerated rhythm should be treated with either antiarrhythmic drugs (Chap.
230), removal of an offending drug, or correction of the metabolic abnormality
or ischemia.
Intracardiac Electrocardiographic Recordings in
Diagnosis and Management
The
main therapeutic decision in patients with AV conduction disturbance is whether
or not a permanent pacemaker is required, and a number of circumstances exist
in which His bundle electrocardiography can be a useful diagnostic tool upon
which to base this decision. It is unquestionable that patients with symptomatic
second- or third-degree AV block should be paced, and therefore, these patients
do not require electrophysiologic study. However, intracardiac ECG recordings
can be useful in at least the following four groups of patients:
Patients with syncope and bundle branch or bifascicular
block without documentation of AV block. In such patients, the demonstration of marked
infra-His bundle conduction disturbances, i.e., a prolonged HV interval
(>100 ms), may usually be taken as an indication of the need for the
insertion of the permanent pacemaker. Complete electrophysiologic evaluation,
including atrial and ventricular programmed stimulation, is indicated to help
identify other possible cardiac etiologies for the syncope. Since the incidence
of significant advanced AV block is low in asymptomatic patients who
have bifascicular block, electrophysiologic evaluation or permanent pacemakers
are not cost-effective. In this group, observation appears most reasonable.
Patients with 2:1 AV conduction. Intracardiac
recordings are necessary to ascertain the site of the conduction disturbance
because the typical ECG features of Mobitz type I or Mobitz type II block
cannot be discerned during a 2:1 pattern of AV conduction on the surface ECG.
Intracardiac recordings may demonstrate that AV nodal block, intra-His bundle
block, infra-His bundle block, or combinations of block may be responsible
(Figs. 229-7 and 229-8). A surface ECG finding that suggests an infra-His
bundle lesion is the presence of alternating bundle branch block associated
with changing PR intervals. Intracardiac recordings in such patients confirm
that the block is almost always in the His-Purkinje system. Increasing block
with exercise or following atropine suggests intra- or infra-His block (Table
229-2). The finding of infra- or intra-His bundle block in patients with
asymptomatic second-degree AV block mandates pacemaker therapy because of the
high likelihood of the development of symptomatic high-grade AV block and
syncope.
Table 229-2: Site of 2:1 Atrioventricular Block
|
||||||||||||||||||||||||||||||||||||||
Patients with Wenckebach block in the presence of bundle
branch block.
This situation, particularly when the maximal change in PR interval is ![]() 50
ms, can suggest intra- or infra-His Wenckebach block, in which case a pacemaker
is mandated. Intracardiac recordings are necessary to make this diagnosis.
50
ms, can suggest intra- or infra-His Wenckebach block, in which case a pacemaker
is mandated. Intracardiac recordings are necessary to make this diagnosis.
Asymptomatic patients with third-degree AV block. In such patients,
electrophysiologic studies may be useful in assessing the stability of the
junctional pacemaker. Pacing is indicated when the His bundle escape pacemaker
is shown to be unstable by an inadequate response to exercise, atropine, or
isoproterenol or by a prolonged junctional recovery time following ventricular
pacing.
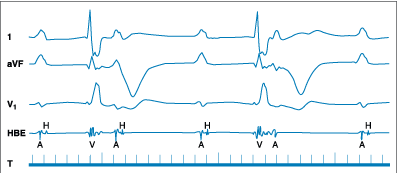
Figure 229-8: Third-degree AV block. The figure shows
surface leads 1, aVF, V1, and an intracardiac His bundle recording (HBE).
Complete heart block is evident on the surface leads. The intracardiac
recording demonstrates an absence of QRS deflection (V) after a His bundle (H)
spike. This indicates block below the His bundle. Note that following the
second QRS complex (V), there is an atrial (A) deflection indicating retrograde
conduction. Retrograde conduction is often present when block is in the
His-Purkinje system but is virtually never present when block is in the AV
node.
![]() Genetics
Genetics
A
number of congenital and familial syndromes involving the cardiac conduction
system have been described. An example of a congenital condition that is
transmitted but not genetic is congenital complete heart block associated with
maternal systemic lupus erythematosus. This disorder is associated with
maternal IgG autoantibodies to several ribonucleoproteins that are
transplacentally transmitted to the fetus and damage the fetal AV node. The
fetal conduction disease is generally clinically evident by the second
trimester and is associated with significant fetal mortality and neonatal
requirement of cardiac pacing.
The
embryonic development of the cardiac septa and conduction system occur
together, and clinical disorders have been described, including the Holt-Oram
syndrome, an autosomal dominant disorder including upper limb dysplasia and
atrial septal defect, often with conduction disturbances in the AV node.
Studies of families with a high incidence of congenital heart disease,
including ostium secundum atrial septal defect and conduction disorders in the
AV node, have identified the gene NKX2-5 on chromosome 5q35 as important in the
regulation of septation and in the development and function of the AV node. A
familial syndrome of progressive complete heart block has also long been
recognized. The gene for this disorder has been mapped to a region on
chromosome 19q13. Familial disorders of SA node function have also been described,
but specific details of abnormal genetic sites are not available.
![]() Treatment
Treatment
Pharmacologic Therapy
Pharmacologic
therapy is usually reserved for acute situations. Atropine (0.5 to 2.0 mg
intravenously) and isoproterenol (1 to 4 ![]() g/min
intravenously) are useful in increasing heart rate and decreasing symptoms in
patients with sinus bradycardia or AV block localized to the AV node. They have
an insignificant effect on lower pacemakers. In patients with neurovascular
syncope, beta blockers and disopyramide have been suggested as methods to
depress left ventricular function and decrease mechanoreceptor-related
reflexes. Mineralocorticoids, ephedrine, and theophylline have also been
reported to be of benefit to occasional patients. Unfortunately, no controlled study
has shown that any of these pharmacologic modalities works in a predictable
fashion in all patients. Further work on delineating different mechanisms in
different patient groups may allow us to apply pharmacologic agents more
appropriately. Long-term therapy of bradyarrhythmias is best accomplished by
pacemakers.
g/min
intravenously) are useful in increasing heart rate and decreasing symptoms in
patients with sinus bradycardia or AV block localized to the AV node. They have
an insignificant effect on lower pacemakers. In patients with neurovascular
syncope, beta blockers and disopyramide have been suggested as methods to
depress left ventricular function and decrease mechanoreceptor-related
reflexes. Mineralocorticoids, ephedrine, and theophylline have also been
reported to be of benefit to occasional patients. Unfortunately, no controlled study
has shown that any of these pharmacologic modalities works in a predictable
fashion in all patients. Further work on delineating different mechanisms in
different patient groups may allow us to apply pharmacologic agents more
appropriately. Long-term therapy of bradyarrhythmias is best accomplished by
pacemakers.
Pacemakers
External
energy sources can be used to stimulate the heart when disorders in impulse
formation and/or transmission lead to symptomatic bradyarrhythmias. Pacer
stimuli can be applied to the atria and/or ventricles. Indications for
pacemaker insertion are listed in the guidelines summarized in Table 229-4.
Temporary Pacing
This
is usually instituted to provide immediate stabilization prior to permanent
pacemaker placement or to provide pacemaker support when a bradycardia is
precipitated by what is presumed to be a transient event such as ischemia or
drug toxicity. Temporary pacing is usually achieved by the transvenous
insertion of an electrode catheter with the catheter positioned in the right
ventricular apex and attached to an external generator. This procedure is
associated with a small risk of cardiac perforation, infection at the insertion
site, and thromboembolism; the risk of the latter two complications increases
markedly if the pacing wire is left in place for more than 48 h. The
development of an entirely external transthoracic cardiac pacing system may
preclude the need for transvenous pacing in selected patients. However,
occasional failure of ventricular capture and significant discomfort related to
the large current required for effective transthoracic ventricular stimulation
preclude the uniform use of this approach.
Permanent Pacing
This
mode of pacing is instituted for persistent or intermittent symptomatic
bradycardia not related to a self-limiting precipitating factor or for
documented infranodal second- or third-degree AV block. Permanent pacing leads
are usually inserted transvenously through the subclavian or cephalic vein with
the leads positioned in the right atrial appendage for atrial pacing and the
right ventricular apex for ventricular pacing. The leads are then attached to
the pulse generator, which is inserted into a subcutaneous pocket below the
clavicle. Epicardial lead placement is used when (1) transvenous access cannot
be obtained; (2) the chest is already open, i.e., in the course of a cardiac
operation; and (3) adequate endocardial lead placement cannot be achieved. Most
pacemaker generators are powered by lithium batteries. The life expectancy of
the generator is related to (1) voltage output required for capture, (2)
requirement for incessant or intermittent pacing, and (3) number of cardiac
chambers paced. Life expectancy of the simple ventricular demand pacemaker can
exceed 10 years.
Pacing Code
A
code consisting of three to five letters has been developed for describing
pacemaker type and function (Table 229-3). The first letter indicates the
chamber(s) paced and is designated V for ventricular pacing, A
for atrial pacing, or D for dual-chamber (both atrial and ventricular)
pacing. The second letter indicates the chamber in which electrical activity is
sensed and is also indicated by A, V, or D. An additional
designation, O, has been used when pacemaker discharge is not dependent
on a sensed electrical activity. The third letter refers to the response to a
sensed electric signal. The letter O represents no response to an
underlying electric signal, usually related to the absence of associated
sensing function; I represents inhibition of pacing function; T
represents triggering of pacing function; and D indicates a dual
response, i.e., spontaneous atrial and ventricular activity inhibiting atrial
and ventricular pacing and atrial activity triggering a ventricular response.
Additional fourth and fifth letters of the pacing code have been recommended to
indicate whether the pacemaker is programmable and has rate modulation (fourth)
and whether special antitachycardia functions are available (i.e.,
antitachycardia pacing, T, and delivery of high- or low-energy shocks).
In the fourth category, M represents multiprogrammability and R
represents rate response ("physiologic") pacing. It follows from the
described code that the standard VVIR (ventricular demand pacemaker) paces the
ventricle, senses the ventricle, is inhibited by sensed spontaneous ventricular
activity, and has rate modulation, while the DDDR pulse generator is capable of
sensing and pacing both the atria and ventricles and has a dual response to the
sensed atrial and ventricular activity as described above (Fig. 229-9). Both
pacemakers have rate modulation (R). "Physiologic" pacemakers
use sensors (muscular activity, respiratory rate, temperature, O2
saturation, QT interval, etc.) as methods to allow the pacemaker to increase
the heart rate in response to physiologic demands, i.e., exercise. These
pacemakers are essential when chronotropic incompetence is present and an
increase in heart rate is required to enhance physiologic performance. Studies
have shown that such "physiologic" pacemakers improve exercise
tolerance and relieve symptoms to a greater degree than fixed-rate pacemakers.
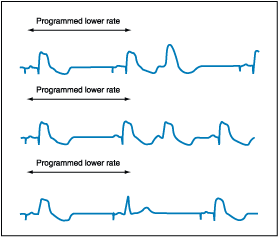
Figure 229-9: Normally functioning DDD pacemaker. All
three panels show a lead II rhythm strip at 50 mm/s. The programmed lower rate
is approximately 55 beats per minute. (Top) AV sequential pacing with a
paced AV interval of 160 ms is shown for the first two complexes. A VPC occurs
and is sensed, resetting the cycle. (Middle) The first beat is AV paced,
but spontaneous sinus P waves and APC trigger a ventricular paced complex with
a sensed P to QRS of 120 ms. (Bottom) After the first AV paced complex, a
paced atrial complex conducts to the ventricle with a PR of 120 ms, inhibiting
the ventricular pacemaker.
Table 229-3: The NASPE/BPEG Generic Pacemaker
Code
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Selection
of the appropriate pacemaker and pacing mode depends on the clinical condition
and the type of bradyarrhythmia being treated. The two most common pacing mode selections
are DDD and VVI. DDD provides AV sequential pacing, which is ideally suited for
the relatively young and active patient who has intact sinus node function or
intermittent dysfunction and high-grade persistent or intermittent AV block.
The DDD mode will allow for physiologic atrial sensed and ventricular paced
rates and improve exercise tolerance. AV synchrony and dual-chamber pacing may
also be desirable in patients with borderline hemodynamic reserve who are
dependent on atrial contribution to cardiac output and in those patients who
develop the pacemaker syndrome (see below) in response to ventricular demand
pacing.
Rate-responsive
DDD (i.e., DDDR) pacing is indicated when chronotropic incompetence is present
in a patient who requires AV synchrony. The DDD pacing mode is contraindicated
in chronic atrial fibrillation or flutter, because rapid and irregular
ventricular pacing will occur to the upper rate limit. In some cases this will
produce a more rapid ventricular rate than the patient's own rate in the
absence of a pacemaker. DDD pacemakers must either automatically switch (i.e.,
mode-switching function) or be reprogrammed to the VVI mode. Almost all such
pacemakers are now combined with some form of rate responsiveness so that when
the device functions in the VVI mode, it also will respond to physiologic
demands (VVIR).
Chronotropic
insufficiency (i.e., the inability of the sinus rate to accelerate) is a
contraindication for a DDD pacemaker, since such a pacemaker will act as a
"fixed-rate" pacemaker at the programmed lower rate. In these
situations, a rate-adaptive or "physiologic" pacemaker is indicated
(VVIR or DDDR). In patients with impaired sinus node function or chronic atrial
fibrillation, a sensor-driven, rate-adaptive pacemaker must be implanted. As
mentioned earlier, these pacemakers automatically adjust ventricular pacing
rates to a sensed indicator of exertion. The DDD pacing mode may also be
contraindicated in patients with intermittent or persistent ventriculoatrial
conduction, who may develop pacemaker-mediated tachycardia (see below).
Programmability of Pacemakers
This
allows for modification of pacing function after implantation and for
adaptation to changes in clinical needs. Pacemaker programming is accomplished
by activation of the programming head positioned over the implanted pulse
generator after making the desired changes in programmable parameters (Table
229-3). A radio frequency system is routinely used to communicate the program
to the pacemaker. A high degree of sophistication is required to recognize the
presence and causes of pacemaker malfunction and their treatment.
Complications
Adverse
effects of permanent pacing are usually associated with failure or malfunction
of the pacing system. These problems are usually secondary to over- or
undersensing, output failure, and/or lead fracture or displacement. Two other
problems may occur. The pacemaker syndrome consists of fatigue,
dizziness, syncope, and distressing pulsations in the neck and chest and can be
associated with adverse hemodynamic effects. The pathophysiologic contributors
to the pacemaker syndrome include (1) loss of atrial contribution to
ventricular systole; (2) vasodepressor reflex initiated by cannon a waves,
which are caused by atrial contractions against a closed tricuspid valve and
observed in the jugular venous pulse (Chap. 225); and (3) systemic and
pulmonary venous regurgitation due to atrial contraction against a closed AV
valve. The symptoms associated with the pacemaker syndrome can be prevented by
maintaining AV synchrony by dual-chamber pacing or, in the case of a
ventricular demand pacemaker, by programming an escape rate 15 to 20 beats per
minute below that of the paced rate (i.e., hysteresis). As a result of this
programming, sinus activity and thus atrial contraction will be less likely to
occur at the same time as ventricular pacing and ventricular contraction. The
second major problem peculiar to dual-chamber pacemakers is the development of pacemaker-mediated
tachycardia. In this instance, retrograde depolarization of the atria,
resulting from a premature ventricular depolarization or a paced ventricular
complex, is sensed and leads to subsequent triggering of ventricular pacing.
This, in turn, can result in repetition of the phenomenon of ventriculoatrial
conduction with the development of an endless-loop, pacemaker-mediated
tachycardia. It may be corrected by reprogramming the atrial refractory period.
The Tachyarrhythmias
Mechanisms
of Tachyarrhythmias
Tachyarrhythmias
may be divided into disorders of impulse propagation and disorders of impulse
formation.
Reentry
Disorders
of impulse propagation (reentry) are generally considered to be the most common
mechanism of sustained paroxysmal tachyarrhythmia. The requirements for
initiating reentry include (1) electrophysiologic inhomogeneity (i.e.,
differences in conduction and/or refractoriness) in two or more regions of the
heart connected with each other to form a potentially closed loop; (2)
unidirectional block in one pathway; (3) slow conduction over an alternative
pathway, allowing time for the initially blocked pathway to recover
excitability; and (4) reexcitation of the initially blocked pathway to complete
a loop of activation (Fig. 230-1). Repetitive circulation of the impulse over
this loop can produce a sustained tachyarrhythmia. While anatomic obstacles may
underlie reentry and provide an inexcitable center around which the impulse can
circulate, they are not essential. Reentrant arrhythmias can be reproducibly
initiated and terminated by premature complexes and rapid stimulation. The
response of these arrhythmias to stimulation can help distinguish them from
arrhythmias caused by triggered activity.
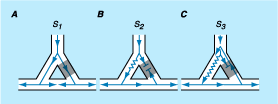
Figure 230-1: Schema of reentry. Y branching of the
Purkinje system to ventricular muscle is shown in panels A through C.
The right limb (gray area) of the Purkinje system has a longer
refractory period than the left. A. During a slow stimulated rate (S1),
conduction proceeds normally over both Purkinje fibers, resulting in collision
in the ventricular muscle. B. An early premature stimulus (S2)
results in block in the Purkinje fiber on the right and slow conduction down
the left. The impulse conducts through the ventricle and attempts to reenter
the initial site of block but fails because this site has not fully recovered
excitability. C. An earlier stimulus (S3) again results in
block on the left. The resulting slower propagation down the left fiber
provides enough time for the initial site of block to recur and allows the
impulse to conduct through it to produce a reentrant circuit.
Enhanced Automaticity
Disorders
of impulse formation can be subdivided into tachyarrhythmias caused by enhanced
automaticity and those caused by triggered activity. In addition to the sinus
node, automatic pacemaker activity can be observed in specialized atrial
fibers, fibers of the atrioventricular (AV) junction, and Purkinje fibers
(Chap. 229). Myocardial cells do not normally possess pacemaker activity.
Enhancement of normal automaticity in latent pacemaker fibers or the
development of abnormal automaticity due to partial depolarization of the
resting membrane occurs as a consequence of a variety of pathophysiologic
states, which include (1) increased endogenous or exogenous catecholamines, (2)
electrolyte disturbances (e.g., hyperkalemia), (3) hypoxia or ischemia, (4)
mechanical effects (e.g., stretch), and (5) drugs (e.g., digitalis).
Tachycardia caused by automaticity cannot be started or stopped by pacing.
Triggered Activity
Rhythms
due to triggered activity are events that do not occur spontaneously but
require a change in cardiac electrical frequency as a trigger. Triggered
activity may be caused by early afterdepolarizations, which occur during phases
2 and 3 of the action potential, or delayed afterdepolarizations, which occur
following completion of phase 3 of the action potential (Fig. 229-2). Triggered
activity has been observed in atrial, ventricular, and His-Purkinje tissue
under conditions such as increased local catecholamine concentration,
hyperkalemia, hypercalcemia, and digitalis intoxication (delayed afterdepolarizations)
or during bradycardia, hypokalemia, or other situations prolonging action
potential duration (early afterdepolarizations). All of these conditions
produce an accumulation of intracellular calcium. With increasing amplitude of
the afterdepolarizations, threshold can be reached and repetitive activity
produced. The exact role of triggered activity in spontaneous clinical
arrhythmias is unknown, but tachyarrhythmias associated with digitalis
intoxication, accelerated idioventricular rhythm in acute infarction and/or
reperfusion, and exercise-induced ventricular tachycardia (VT) are believed to
be caused by triggered activity due to delayed afterdepolarizations. Torsade
de pointes ("twisting of the points"; polymorphic VT associated
with long QT intervals) may be caused by triggered activity due to early
afterdepolarizations, although reentry may also be operative.
The
use of electrophysiologic studies, i.e., intracardiac recordings and programmed
stimulation, has greatly expanded the understanding of the mechanisms of
tachyarrhythmias. In addition to helping diagnose arrhythmias, these techniques
may be of value in determining the most appropriate types of therapy because
they allow the physician to observe the hemodynamic and symptomatic consequences
of the arrhythmia in the presence or absence of therapy. Electrophysiologic
studies of tachycardias require the positioning of multiple electrode catheters
at critical areas within the heart. These electrodes must be capable of both
stimulating and recording from multiple sites in the atria and/or ventricles.
Premature Complexes
Atrial Premature Complexes
(APC)
APCs
can be found on 24-h Holter monitoring in over 60% of normal adults. APCs are
usually asymptomatic and benign, although at times they may be associated with
palpitations. In susceptible patients, they can initiate paroxysmal
supraventricular tachycardias. APCs may originate from any location in either
atrium, and they are recognized on the electrocardiogram (ECG) as early P waves
with a morphology that differs from the sinus P wave (Fig. 230-2). While APCs
usually conduct to the ventricles when they occur late in the cardiac cycle,
early APCs may reach the AV conduction system while it is still in its relative
refractory period, resulting in a conduction delay manifested by prolonged PR
interval following the premature P wave (Fig. 230-2). Very early APCs may even
block in the AV node if this structure is encountered during its effective
refractory period. APCs, whether conducted or not, are usually followed by a
pause before a return to sinus activity. Most commonly, an APC enters and
resets the sinus node, so the sum of the pre- and postextrasystolic PP
intervals is less than the sum of two sinus PP intervals (Fig. 230-2). In this
case, the pause is said to be less than fully compensatory. The QRS complex
following most APCs is normal, although early APCs may be followed by
aberrantly conducted QRS complexes due to the premature complex falling within
the relative refractory period of the His-Purkinje system.

Figure 230-2: ECG lead II. Sinus rhythm with one atrial
premature complex (arrow). Note the difference in P-wave configuration
between sinus and the premature atrial complexes. In addition, note that the PR
interval of the premature complex is prolonged, due to slowed conduction of the
premature impulse through the AV conduction system.
Since
most APCs are asymptomatic, treatment is not required. When they cause
palpitations or trigger paroxysmal supraventricular tachycardias (see below),
treatment may be useful. Factors that precipitate APCs, such as alcohol,
tobacco, or adrenergic stimulants, should be identified and eliminated; in
their absence, mild sedation or the use of a beta blocker may be tried.
AV
Junctional Complexes
The
site of origin of these complexes is thought to be in the bundle of His, since
the normal AV node in vivo possesses no automaticity. AV junctional complexes
are less common than either atrial or ventricular premature complexes and are
more often associated with cardiac disease or digitalis intoxication. Junctional
premature impulses can conduct both antegradely to the ventricles and
retrogradely to the atrium and, on rare occasions, may fail to conduct in
either direction. Premature AV junctional complexes can be recognized by
normal-appearing QRS complexes that are not preceded by a P wave. Retrograde P
waves (inverted in leads II, III, and aVF) may be observed after the QRS
complex.
While
often asymptomatic, junctional premature complexes may be associated with
palpitations and cause cannon a waves, which may result in distressing
pulsations in the neck. When symptomatic, they should be treated like APCs.
VENTRICULAR PREMATURE COMPLEXES (VPCs)
These
are among the most common arrhythmias and occur in patients with and without
heart disease. Of adult males, ![]() 60%
will exhibit VPCs during a 24-h Holter monitoring. In patients without heart
disease, VPCs have not been shown to be associated with any increased incidence
in mortality or morbidity. VPCs may occur in up to 80% of patients with
previous myocardial infarction, and in this setting, if frequent (>10 per
hour) and/or complex (occurring in couplets), they have been associated with
increased mortality. However, cardiac mortality in such patients usually occurs
in association with significantly impaired ventricular function. While frequent
and complex ventricular ectopy is an independent risk factor, it is not as
strong a risk factor as is impaired ventricular function. Moreover, even though
ventricular tachycardia and/or fibrillation may be the basis for the sudden
death in these patients, this does not a priori establish a cause-and-effect
relation between spontaneous ectopy and life-threatening ventricular
tachycardia or fibrillation. Very early cycle (R-on-T) VPCs have been stated by
some to increase the risk of sudden death. Although this has been observed
during acute ischemia and in the setting of QT prolongation, frequently, VT or
fibrillation is precipitated by VPCs that occur after the T wave of the prior
beat.
60%
will exhibit VPCs during a 24-h Holter monitoring. In patients without heart
disease, VPCs have not been shown to be associated with any increased incidence
in mortality or morbidity. VPCs may occur in up to 80% of patients with
previous myocardial infarction, and in this setting, if frequent (>10 per
hour) and/or complex (occurring in couplets), they have been associated with
increased mortality. However, cardiac mortality in such patients usually occurs
in association with significantly impaired ventricular function. While frequent
and complex ventricular ectopy is an independent risk factor, it is not as
strong a risk factor as is impaired ventricular function. Moreover, even though
ventricular tachycardia and/or fibrillation may be the basis for the sudden
death in these patients, this does not a priori establish a cause-and-effect
relation between spontaneous ectopy and life-threatening ventricular
tachycardia or fibrillation. Very early cycle (R-on-T) VPCs have been stated by
some to increase the risk of sudden death. Although this has been observed
during acute ischemia and in the setting of QT prolongation, frequently, VT or
fibrillation is precipitated by VPCs that occur after the T wave of the prior
beat.
VPCs
are recognized by wide (usually >0.14 s), bizarre QRS complexes that are not
preceded by P waves (Fig. 230-3A). They may bear a relatively fixed
relationship to the preceding sinus complex (i.e., fixed coupled VPCs). When
fixed coupling is not present and the interval between VPCs has a common
denominator, ventricular parasystole is said to be present (Fig. 230-4).
Under these circumstances, the VPCs are a manifestation of abnormal
automaticity of a protected ventricular focus. Because this focus is not
penetrated by sinus impulses, it is not reset by them, and the interectopic
intervals remain relatively fixed (![]() 120
ms variation of mean RR cycle length).
120
ms variation of mean RR cycle length).
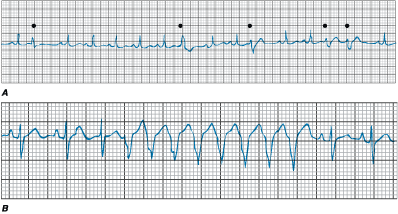
Figure 230-3: A. Single ventricular
ectopy. During sinus rhythm, five premature ventricular complexes (filled
circles) occur. Note that the QRS configuration is bizarre and different from
that during sinus rhythm. The premature ventricular complexes are not preceded
by P waves. The QRS widths of the premature complexes are 120-160 ms and
multiple morphologies are present. The pause following the premature complexes
is fully compensatory, the sinus beat after the premature complex occurring on time.
B. Nonsustained ventricular tachycardia (VT). Following two sinus beats,
an atrial premature contraction with long PR interval initiates an 8-beat run
of wide complex tachycardia. Atrial activity can be seen following the fourth
and seventh beats. The greater number of QRS complexes compared with P waves
confirms the diagnosis of VT.
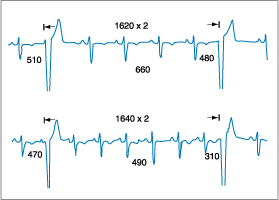
Figure 230-4: Ventricular parasystole. At varying sinus
cycle lengths during exercise, interectopic intervals remain constant at 1620
to 1640 ms. However, the coupling intervals between sinus and ectopic complexes
vary between 510 and 310 ms.
VPCs
may occur singly; in patterns of bigeminy, in which every sinus beat is
followed by a VPC; in trigeminy, in which two sinus beats are followed by a
VPC; in quadrigeminy, etc. Two successive VPCs are termed pairs or couplets,
while three or more consecutive VPCs are termed ventricular tachycardia
when the rate exceeds 100 beats per minute (Fig. 230-3B). VPCs may have
similar morphologies (monomorphic, or uniform) or different morphologies
(polymorphic, or multiformed).
Most
commonly, VPCs are not conducted retrogradely to the atrium to reset the
sinoatrial node. Thus they produce a fully compensatory pause; i.e., the
interval between conducted sinus beats that bracket the VPC equals two basic RR
intervals. Ventricular impulses may also manifest retrograde conduction to the
atrium and cause inverted P waves in leads II, III, and aVF. This retrograde
atrial activation can reset the sinus node, and the pause that results may
therefore be less than compensatory. In many instances, the VPC will not be
associated with retrograde ventriculoatrial (VA) conduction but may block
retrogradely in the AV node. This renders the AV node refractory to the
subsequent sinus beat and causes slowed conduction (i.e., prolonged PR
interval) or block of the next sinus P wave. This prolonged PR interval is said
to be a manifestation of concealed retrograde conduction of the ventricular
impulse into the AV node. A VPC that does not produce any manifestation of
retrograde concealed conduction and fails to influence the oncoming sinus
impulse is termed an interpolated VPC.
VPCs
can cause palpitations or neck pulsations secondary to either the occurrence of
cannon a waves or the increased force of contraction due to
postextrasystolic potentiation of ventricular contractility. Patients with
frequent VPCs or bigeminy may rarely develop syncope or lightheadedness because
the VPCs do not result in an adequate stroke volume and the cardiac output is
reduced by the "halving" of the heart rate.
![]() Treatment
Treatment
In
the absence of cardiac disease, isolated asymptomatic VPCs, regardless of
configuration and frequency, need no treatment. When arrhythmias are
symptomatic, the symptoms should first be addressed by either allaying the
patient's anxiety or, if this is not successful, reducing the frequency of the
VPCs with antiarrhythmic agents. ![]() -Adrenergic
blockers may be successful in managing VPCs that occur primarily in the daytime
or under stressful situations and in specific settings such as mitral valve prolapse
and thyrotoxicosis. While other antiarrhythmic agents may be tried should this
be unsuccessful, their risk may outweigh any benefits. In patients with cardiac
disease, frequent VPCs are associated with an increased risk of sudden and
nonsudden cardiac death, and many physicians have attempted to eliminate or
reduce the frequency of these VPCs in an attempt to reduce this risk. However,
the cause-and-effect relationship of the VPCs to fatal events has never been
established. The ability of pharmacologic antiarrhythmic therapy guided by
continuous ECG monitoring to reduce the risk of sudden death in postmyocardial
infarction patients with frequent (
-Adrenergic
blockers may be successful in managing VPCs that occur primarily in the daytime
or under stressful situations and in specific settings such as mitral valve prolapse
and thyrotoxicosis. While other antiarrhythmic agents may be tried should this
be unsuccessful, their risk may outweigh any benefits. In patients with cardiac
disease, frequent VPCs are associated with an increased risk of sudden and
nonsudden cardiac death, and many physicians have attempted to eliminate or
reduce the frequency of these VPCs in an attempt to reduce this risk. However,
the cause-and-effect relationship of the VPCs to fatal events has never been
established. The ability of pharmacologic antiarrhythmic therapy guided by
continuous ECG monitoring to reduce the risk of sudden death in postmyocardial
infarction patients with frequent (![]() 6
per minute) VPCs was tested by the Cardiac Arrhythmia Suppression Trial (CAST).
This study compared mortality in patients whose ectopy was suppressed by one of
three agents (encainide, flecainide, or moricizine) and then randomized to
treat with either the "effective" drug or placebo. After a mean
follow-up of 2 years, the study was discontinued because both the sudden death
and overall mortality rate were significantly increased in patients receiving
antiarrhythmic agents. This study has shown that in patients having the
characteristics of the study population, abolition of ventricular ectopy by
pharmacologic therapy cannot be used as a marker to define reduction of the
risk of sudden death after myocardial infarction and, in fact, may increase
mortality. Recent studies have evaluated the use of electrophysiologic testing
and implantable cardioverter/defibrillator (ICD) placement in the management of
patients at high risk for sudden death (i.e., those with left ventricular
ejection fractions <40% and nonsustained VT). These studies have found that
induction of a sustained ventricular arrhythmia through programmed electrical
stimulation selects a group of these patients whose prognosis is improved with
implantation of a defibrillator. These studies have found no correlation
between the rate, morphology, or duration of nonsustained episodes of VT and
the likelihood of having a sustained ventricular arrhythmia.
6
per minute) VPCs was tested by the Cardiac Arrhythmia Suppression Trial (CAST).
This study compared mortality in patients whose ectopy was suppressed by one of
three agents (encainide, flecainide, or moricizine) and then randomized to
treat with either the "effective" drug or placebo. After a mean
follow-up of 2 years, the study was discontinued because both the sudden death
and overall mortality rate were significantly increased in patients receiving
antiarrhythmic agents. This study has shown that in patients having the
characteristics of the study population, abolition of ventricular ectopy by
pharmacologic therapy cannot be used as a marker to define reduction of the
risk of sudden death after myocardial infarction and, in fact, may increase
mortality. Recent studies have evaluated the use of electrophysiologic testing
and implantable cardioverter/defibrillator (ICD) placement in the management of
patients at high risk for sudden death (i.e., those with left ventricular
ejection fractions <40% and nonsustained VT). These studies have found that
induction of a sustained ventricular arrhythmia through programmed electrical
stimulation selects a group of these patients whose prognosis is improved with
implantation of a defibrillator. These studies have found no correlation
between the rate, morphology, or duration of nonsustained episodes of VT and
the likelihood of having a sustained ventricular arrhythmia.
Antiarrhythmic
agents can also produce the lethal arrhythmias that they are given to prevent
(proarrhythmic effects). Thus therapy directed toward VPCs in the setting of
chronic cardiac disease may result in an inappropriate and costly use of agents
without proven efficacy and with potential side effects in many patients. The
high incidence of side effects and the frequent exacerbation of arrhythmias
caused by all antiarrhythmic drugs make it mandatory to monitor patients being
treated with such agents.
In
acute myocardial infarction, the greatest incidence of primary ventricular
fibrillation occurs within the first 24 h (Chap. 243). Temporary prophylactic
antiarrhythmic therapy with lidocaine or procainamide was formerly recommended
for all patients with acute infarction, regardless of the presence or degree of
spontaneous ectopy. However, failure to improve overall survival and drug
toxicity have led most physicians to recommend prophylactic antiarrhythmic
therapy only to young patients with complicated infarctions, where a favorable
risk-benefit ratio may be obtained. Other studies have shown that intravenous
beta blockers may also reduce the incidence of primary ventricular
fibrillation.
Tachycardias
Tachycardias refer to arrhythmias with three or more
complexes at rates exceeding 100 beats per minute; they occur more often in
structurally diseased than in normal hearts. Those paroxysmal tachycardias that
are initiated by APCs or VPCs are considered to be due to reentry, except some
of the digitalis-induced tachyarrhythmias, which are probably due to triggered
activity (see below).
If
the patient is hemodynamically stable, an attempt should be made to determine
the mechanism and origin of the tachycardia, since this will usually lead to an
appropriate therapeutic decision. Information to be obtained from the ECG
includes (1) the presence, frequency, morphology, and regularity of P waves and
QRS complexes; (2) the relationship between atrial and ventricular activity;
(3) a comparison of the QRS morphology during sinus rhythm and during the
tachycardia; and (4) the response to carotid sinus massage or other vagal
maneuvers. It is useful first to compare a 12-lead ECG during the tachycardia
with one recorded during sinus rhythm. One can also utilize the electrodes
situated at the end of a flexible pacing catheter inserted into the esophagus
behind the left atrium to record atrial activity.
Observation
of the jugular venous pulse can provide clues to the presence of atrial
activity and its relationship to ventricular ectopy. Intermittent cannon a
waves suggest AV dissociation, while persistent cannon a waves suggest 1:1 VA
conduction. Flutter waves may be seen or no atrial activity may be apparent, as
in the presence of atrial flutter and fibrillation, respectively. The arterial
pulse may also manifest AV dissociation or atrial fibrillation by demonstrating
variations in amplitude. A first heart sound of variable intensity during a
regular rhythm also suggests AV dissociation or atrial fibrillation (AF).
Carotid
sinus pressure should only be applied while the patient is
electrocardiographically monitored with resuscitative equipment available to
manage the rare episode of asystole and/or ventricular fibrillation associated
with this procedure. Carotid sinus massage should not be performed in patients
with carotid arterial bruits. The patient should be positioned flat with the
neck extended. Massage of one carotid bulb at a time should be performed by
applying firm pressure just underneath the angle of the jaw for up to 5 s.
Alternative vagomimetic maneuvers include the Valsalva maneuver, immersion of
the face in cold water, and administration of 5 to 10 mg edrophonium.
Sinus Tachycardia
In
the adult, sinus tachycardia is said to be present when the heart rate exceeds
100 beats per minute (bpm): sinus tachycardia rarely exceeds 200 bpm and is not
a primary arrhythmia; instead, it represents a physiologic response to a
variety of stresses, such as fever, volume depletion, anxiety, exercise,
thyrotoxicosis, hypoxemia, hypotension, or congestive heart failure. Sinus
tachycardia has a gradual onset and offset. The ECG demonstrates P waves with
sinus contour preceding each QRS complex. Carotid sinus pressure usually
produces modest slowing with a gradual return to the previous rate upon
cessation. This contrasts with the response of paroxysmal supraventricular
tachycardias, which may slow slightly and terminate abruptly.
![]() Treatment
Treatment
Sinus
tachycardia should not be treated as a primary arrhythmia, since it is almost
always a physiologic response to a demand placed on the heart. As such, the
therapy should be directed to the primary disorder. This may involve
institution of digitalis and/or diuretics for heart failure and oxygen for
hypoxemia, treatment of thyrotoxicosis, volume repletion, aspirin for fever, or
tranquilizers for emotional upset.
Atrial Fibrillation
AF
is a common arrhythmia that may occur in paroxysmal and persistent forms. It
may be seen in normal subjects, particularly during emotional stress or
following surgery, exercise, acute alcoholic intoxication, or a prominent surge
of vagal tone (i.e., vasovagal response). It may also occur in patients with
heart or lung disease who develop acute hypoxia, hypercapnia, or metabolic or
hemodynamic derangements. Persistent AF usually occurs in patients with
cardiovascular disease, most commonly rheumatic heart disease, nonrheumatic
mitral valve disease, hypertensive cardiovascular disease, chronic lung
disease, atrial septal defect, and a variety of miscellaneous cardiac
abnormalities. AF may be the presenting finding in thyrotoxicosis. So-called
lone AF, which occurs in patients without underlying heart disease, often
represents the tachycardia phase of the tachycardia-bradycardia syndrome.
The
morbidity associated with AF is related to (1) excessive ventricular rate,
which in turn may lead to hypotension, pulmonary congestion, or angina pectoris
in susceptible individuals; (2) the pause following cessation of AF, which can
cause syncope; (3) systemic embolization, which occurs most commonly in
patients with rheumatic heart disease (Table 230-1); (4) loss of the
contribution of atrial contraction to cardiac output, which may cause fatigue;
and (5) anxiety secondary to palpitations. In patients with severe cardiac
dysfunction, particularly those with hypertrophied, noncompliant ventricles,
the combination of the loss of the atrial contribution to ventricular filling
and the abbreviated filling period due to the rapid ventricular rate in AF can
produce marked hemodynamic instability, resulting in hypotension, syncope, or
heart failure. In patients with mitral stenosis, in whom ventricular filling
time is critical, development of AF with a rapid ventricular rate may
precipitate pulmonary edema (Chap. 236). AF may also cause a cardiomyopathy
related to persistent rapid rates (so-called tachycardia-induced
cardiomyopathy).
Table 230-1: Factors Associated with High Risk
of Stroke in Patients with Atrial Fibrillation
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||
AF
is characterized by disorganized atrial activity without discrete P waves on
the surface ECG (Fig. 230-5A). Atrial activation is manifested by an
undulating baseline or by more sharply inscribed atrial deflections of varying
amplitude and frequency ranging from 350 to 600 beats per minute. The
ventricular response is irregularly irregular. This results from the large
number of atrial impulses that penetrate the AV node, making it partially
refractory to subsequent impulses. This effect of nonconducted atrial impulses
to influence the response to subsequent atrial impulses is termed concealed
conduction. As a result, the ventricular response is relatively slow,
considering the actual atrial rate. AF may convert to atrial flutter,
especially in response to antiarrhythmic drugs like quinidine or flecainide. If
AF converts to atrial flutter, which has a slower atrial rate, the effect of
concealed conduction may be diminished, and a paradoxic increase in the
ventricular response may occur. The main factor determining the rate of the
ventricular response is the functional refractory period of the AV node or the
most rapid paced rate at which 1:1 conduction through the AV node can be
observed.
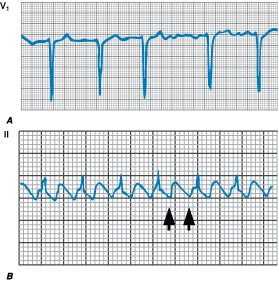
Figure 230-5: Atrial fibrillation and atrial flutter. A.
Lead V1 demonstrating an irregular ventricular rhythm associated
with poorly defined irregular atrial activity consistent with atrial
fibrillation. B. Lead II demonstrates atrial flutter, identified by the
regular "sawtooth-like" activity (arrows) at an atrial rate of
300 bpm with 2:1 ventricular response.
If,
in the presence of AF, the ventricular rhythm becomes regular and slow (e.g.,
30 to 60 bpm), complete heart block is suggested, and if the ventricular rhythm
is regular and rapid (e.g., ![]() 100
bpm), a tachycardia arising in the AV junction or ventricle should be
suspected. Digitalis intoxication is a common cause of both phenomena.
100
bpm), a tachycardia arising in the AV junction or ventricle should be
suspected. Digitalis intoxication is a common cause of both phenomena.
Patients
with AF exhibit a loss of a waves in the jugular venous pulse and
variable pulse pressures in the carotid arterial pulse. The first heart sound
usually varies in intensity. On echocardiography, the left atrium is frequently
enlarged, and in patients in whom the left atrial diameter exceeds 4.5 cm, it may
be difficult to convert AF to sinus rhythm and/or maintain the latter, despite
therapy.
![]() Treatment
Treatment
In
acute AF, a precipitating factor such as fever, pneumonia, alcoholic
intoxication, thyrotoxicosis, pulmonary emboli, congestive heart failure, or
pericarditis should be sought. When such a factor is present, therapy should be
directed toward the primary abnormality. If the patient's clinical status is
severely compromised, electrical cardioversion is the treatment of choice. In
the absence of severe cardiovascular compromise, slowing of ventricular rate
becomes the initial therapeutic goal. This may be most rapidly accomplished
with ![]() -adrenergic
blockers and/or calcium channel antagonists. Both prolong the refractory period
of the AV node and slow conduction within it. When catecholamine levels or
sympathetic nervous system tone is likely to be elevated, beta blockers may be
favored. Digitalis preparations are less effective, take longer to act, and are
associated with more toxicity. Conversion to sinus rhythm may then be
attempted. Prior to cardioversion, precautions must be taken to reduce the risk
of systemic embolization. Patients should be anticoagulated to an INR of at
least 1.8 for the prior 3 consecutive weeks or have had AF for <48 h.
Alternatively, for those patients with AF for >48 h who are not
anticoagulated, a transesophageal echocardiogram can exclude the presence of
left atrial thrombus and allow safe cardioversion. Following cardioversion,
anticoagulation must be maintained for at least 4 weeks until atrial mechanical
function returns to normal.
-adrenergic
blockers and/or calcium channel antagonists. Both prolong the refractory period
of the AV node and slow conduction within it. When catecholamine levels or
sympathetic nervous system tone is likely to be elevated, beta blockers may be
favored. Digitalis preparations are less effective, take longer to act, and are
associated with more toxicity. Conversion to sinus rhythm may then be
attempted. Prior to cardioversion, precautions must be taken to reduce the risk
of systemic embolization. Patients should be anticoagulated to an INR of at
least 1.8 for the prior 3 consecutive weeks or have had AF for <48 h.
Alternatively, for those patients with AF for >48 h who are not
anticoagulated, a transesophageal echocardiogram can exclude the presence of
left atrial thrombus and allow safe cardioversion. Following cardioversion,
anticoagulation must be maintained for at least 4 weeks until atrial mechanical
function returns to normal.
Antiarrhythmic
medications in either oral or intravenous form may be employed but are only
modestly effective in restoring sinus rhythm. When antiarrhythmic agents such
as the quinidine-like drugs (type 1A) or the flecainide-like agents (type 1C)
are used (Table 230-2), it is important to increase AV node refractoriness
prior to administering such drugs because their vagolytic effect and/or their
ability to convert AF to atrial flutter may reduce the concealed conduction in
the AV node and lead to an excessively rapid ventricular response. ![]() -Adrenergic
blockers are especially useful in this regard.
-Adrenergic
blockers are especially useful in this regard.
Table 230-2: Classification of Antiarrhythmic
Drugs
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Direct-current
electrical cardioversion is a highly effective method to restore sinus rhythm, either
as a primary method of therapy or following the failure of antiarrhythmic
medications. Electrical cardioversion is accomplished through the delivery of
at least 200 W · s of energy between electrodes placed to the right of the
sternum and the cardiac apex or to the left of the scapula. If external
cardioversion is unsuccessful, internal cardioversion with energy delivered
between two catheters inside the heart or one inside and a patch outside the
heart may prove effective.
It
is unlikely that patients with chronic AF will convert to and remain in sinus
rhythm in the presence of long-standing rheumatic heart disease and/or when the
atria are markedly enlarged. It is also unlikely for patients with recurrent,
paroxysmal lone AF to be converted to and maintained in sinus rhythm.
The
goal of therapy in patients in whom AF cannot be converted to sinus rhythm is
control of the ventricular response. This can usually be accomplished by
digitalis, beta blockers, or calcium channel blockers singly or in combination.
In occasional patients, the ventricular response cannot be controlled by
pharmacologic therapy alone. In such patients, the creation of complete heart
block by radiofrequency catheter ablation of the AV junction followed by
permanent pacemaker implantation is appropriate. Surgical or direct-current
catheter ablation of the AV junction is rarely required to achieve AV block.
If
sinus rhythm is restored electrically or pharmacologically, quinidine or
related agents as well as the class IC agents (e.g., flecainide), sotalol, or
amiodarone may be used to prevent recurrence. In patients in whom cardioversion
is unsuccessful or in whom AF has recurred or is likely to recur despite
antiarrhythmic therapy, it is probably wisest to allow the patient to remain in
AF and to control the ventricular response with calcium antagonists, ![]() -adrenergic
blockers, or digitalis glycosides. Since such patients are always at risk of
systemic embolization, particularly in the presence of organic heart disease,
chronic anticoagulation must be considered (Table 230-3). Chronic
anticoagulation is particularly important in the elderly, where the attributable
risk of AF for stroke approaches 30%. Several studies have now demonstrated
conclusively that the incidence of embolization in patients with AF not
associated with valvular heart disease is reduced by chronic anticoagulation
with warfarin-like agents. Aspirin also may be effective for this purpose in
patients who are not at high risk for stroke. Although anticoagulation may be
associated with hemorrhagic complications, the risk is largely associated with
INRs above the recommended range of 1.8 to 3.0. Recommendations for the
selection of antiarrhythmic medications to prevent the recurrence of AF are
shown in Fig. 230-6.
-adrenergic
blockers, or digitalis glycosides. Since such patients are always at risk of
systemic embolization, particularly in the presence of organic heart disease,
chronic anticoagulation must be considered (Table 230-3). Chronic
anticoagulation is particularly important in the elderly, where the attributable
risk of AF for stroke approaches 30%. Several studies have now demonstrated
conclusively that the incidence of embolization in patients with AF not
associated with valvular heart disease is reduced by chronic anticoagulation
with warfarin-like agents. Aspirin also may be effective for this purpose in
patients who are not at high risk for stroke. Although anticoagulation may be
associated with hemorrhagic complications, the risk is largely associated with
INRs above the recommended range of 1.8 to 3.0. Recommendations for the
selection of antiarrhythmic medications to prevent the recurrence of AF are
shown in Fig. 230-6.
Figure 230-6: Recommendations for the selection of
antiarrhythmic medications to prevent the recurrence of atrial fibrillation.
See Tables 230-2 and 230-4 for definition of types IA and IC drugs. An
atrioventricular nodal blocking agent (i.e., beta blocker, calcium channel
blocker, or digoxin) should be added to all type IC and IA agents as well as to
dofetilide. LVEF, left ventricular ejection fraction; CHF, congestive heart
failure; CAD, coronary artery disease; EF, ejection fraction.
Table 230-3: Recommendations for Long-Term
Anticoagulation in Patients with Chronic Atrial Fibrillation
|
||||||||||||||||||||||||||||||||||||||||||||||||
Ablation therapy for cure of AF is an active area of investigation.
This therapy is particularly attractive for the small subset of patients who
have a focal atrial tachycardia that degenerates into AF. These automatic foci
are often located in the pulmonary veins, and a targeted ablation in these
areas may be curative. While ablation of these foci is possible, the procedure
can result in pulmonary vein stenosis, pulmonary hypertension, and stroke.
Further technologic advances are necessary before this procedure can be more
widely and safely performed. A more morbid approach involves making multiple
lesions in the right and left atria (MAZE procedure) to compartmentalize the
electrical conductance of these chambers and disallow the propagation of
fibrillatory waves. The morbidity, mortality, and success rate of such
catheter-based procedures renders them experimental at this time.
Atrial Flutter
This
arrhythmia occurs most often in patients with organic heart disease. Flutter
may be paroxysmal, in which case there is usually a precipitating factor, such as
pericarditis or acute respiratory failure, or it may be persistent. Atrial
flutter (as well as AF) is very common during the first week following
open-heart surgery. Atrial flutter is usually less long-lived than is AF,
although on occasion it may persist for months to years. Most commonly, if it
lasts for more than a week, atrial flutter will convert to AF. Systemic
embolization is less common in atrial flutter than in AF.
Atrial
flutter is characterized by an atrial rate between 250 and 350 bpm. Typically,
the ventricular rate is half the atrial rate, i.e., approximately 150 bpm. If
the atrial rate is slowed to <220 beats per minute by antiarrhythmic agents
such as quinidine, which also possess vagolytic properties, the ventricular
rate may rise suddenly because of the development of 1:1 AV conduction.
Classically, flutter waves are seen as regular sawtooth-like atrial activity,
most prominent in the inferior leads (Fig. 230-5B). When the ventricular
response is regular and not a simple fraction of the atrial rate, complete AV
block is present, which may be a manifestation of digitalis toxicity.
Activation mapping suggests that atrial flutter is a form of atrial reentry
localized to the right atrium.
![]() Treatment
Treatment
The
most effective treatment of atrial flutter is direct-current cardioversion,
which can be accomplished at low energy (25 to 50 W · s) under mild sedation.
Higher energies (100 to 200 W · s) are often used because they are less likely
to cause AF, which not infrequently occurs following lower energy delivery.
Although atrial flutter is associated with a slightly lower risk of embolization
than AF, the same precautions should be followed in regard to anticoagulation
as are used with AF. In patients who develop atrial flutter following
open-heart surgery or recurrent flutter in the setting of acute myocardial
infarction, particularly if they are being treated with digitalis, atrial
pacing (using temporary pacing wires implanted at the time of operation or a
pacing lead inserted into the atrium pervenously) at rates of 115 to 130% of
the atrial flutter rate can usually convert the atrial flutter to sinus rhythm.
Atrial pacing may also result in the conversion of atrial flutter to AF, which
allows for easier control of the ventricular response. If immediate conversion
of atrial flutter is not mandated by the patient's clinical status, the
ventricular response should first be slowed by blocking the AV node with a beta
blocker, calcium antagonist, or digitalis. Digitalis is the least effective and
occasionally converts atrial flutter into AF. Once AV nodal conduction is
slowed with any of these drugs, an attempt to convert flutter to sinus rhythm
using a class I (A or C) agent or amiodarone should be made. Increasing doses
of the drug selected are administered until the rhythm converts or side effects
occur. Ibutilide is a new antiarrhythmic agent that is administered
intravenously and appears to be particularly effective for conversion of atrial
flutter to sinus rhythm.
Quinidine,
other Class IA drugs, flecainide, propafenone, sotalol, and amiodarone (Table
230-4) may be useful in preventing recurrences of atrial flutter.
Radiofrequency ablation is a highly effective treatment for patients with the
most typical forms of atrial flutter, which are due to reentry around the
tricuspid valve in a counterclockwise or clockwise fashion. The coronary sinus
and inferior vena cava cause the wavefront of activation to pass between them
and the tricuspid valve. Ablation of the narrowed isthmus using radiofrequency
energy can cure flutter in >85% of cases.
Table 230-4: Drugs Used to Treat Cardiac
Tachyarrhythmias
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Paroxysmal Supraventricular Tachycardias (PSVT)
In
most cases, functional differences in conduction and refractoriness in the AV
node or the presence of an AV bypass tract provide the substrate for the
development of PSVT (previously termed paroxysmal atrial tachycardia).
Electrophysiologic studies have demonstrated that reentry is responsible for
the vast majority of cases of PSVT (Fig. 230-7). Reentry has been localized to
the sinus node, atrium, AV node, or a macroreentrant circuit involving
conduction in the antegrade direction through the AV node and retrograde
through an AV bypass tract. Such a bypass tract may also conduct antegradely,
in which case the Wolff-Parkinson-White (WPW) syndrome is said to be present.
When the bypass tract manifests only retrograde conduction, it is termed a concealed
bypass tract (Fig. 230-7B). In these cases, the QRS complex during
sinus rhythm is normal. In the absence of the WPW syndrome, reentry through the
AV node or through a concealed bypass tract makes up more than 90% of all
PSVTs.
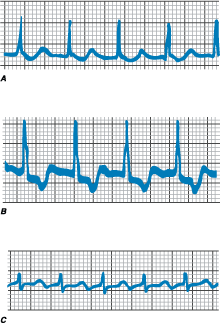
Figure 230-7: Examples of reentrant supraventricular
tachycardia (SVT). A. AV nodal reentry. No P waves are visible. B.
AV reentry using a concealed bypass tract. Inverted retrograde P waves are
superimposed on the T wave. C. Intraatrial reentry. Inverted P waves
follow the T waves and precede the QRS complex.
AV
Nodal Reentrant Tachycardia
There
is no age or disease predisposition for the development of AV nodal reentrant
tachycardia, the most common cause of supraventricular tachycardia. It is, however,
more commonly observed in women. It usually presents as a regular narrow QRS
complex tachycardia at rates of 120 to 250 bpm. APCs that initiate the
arrhythmia are almost always associated with a prolonged PR interval.
Retrograde P waves may be absent, buried in the QRS complex, or appear as
distortions at the terminal parts of the QRS complex (Fig. 230-7A).
AV
nodal reentrant PSVT (Fig. 230-8) can be reproducibly initiated and terminated
by appropriately timed atrial premature stimuli. The onset of the tachycardia
is almost always associated with prolongation of the PR interval due to marked
AV nodal conduction delay (prolonged AH interval) following the APC that is
critical for the genesis of the arrhythmia. The sudden prolongation of the AH
interval is consistent with the concept of dual AV nodal pathways: a fast
pathway, which exhibits rapid conduction and a long refractory period, and
a slow pathway, which has a short refractory period but conducts slowly.
During sinus rhythm, only conduction over the fast pathway is manifest,
resulting in a normal PR interval (Fig. 230-8). Atrial extrastimuli at a
critical coupling interval are blocked in the fast pathway because of its
longer refractory period and are conducted slowly through the slow pathway. If
conduction down the slow pathway is slow enough to allow the previously
refractory fast pathway time to recover excitability, a single atrial (echo)
reentrant beat or sustained tachycardia ensues. A critical balance between
conduction velocity and refractoriness within the node is required to sustain
AV nodal reentry. Retrograde atrial and antegrade ventricular activation occur
simultaneously, explaining why P waves may not be apparent on the surface ECG.
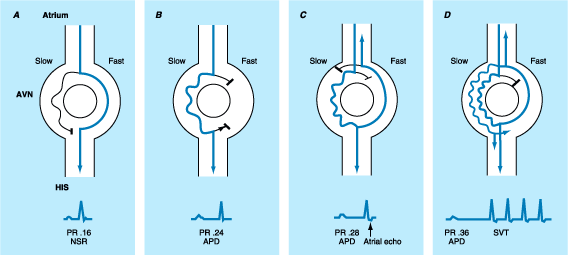
Figure 230-8: Mechanism of AV nodal reentry: The
atrium, AV node (AVN), and His bundle are shown schematically. The AV node is
longitudinally dissociated into two pathways, slow and fast, with different
functional properties (see text). In each panel of this diagram, blue lines
denote excitation in the AV node, which is manifest on the surface
electrocardiogram, while black lines denote conduction, which is concealed and
not apparent on the surface electrocardiogram. A. During sinus rhythm
(NSR) the impulse from the atrium conducts down both pathways. However, only
conduction over the fast pathway is manifest on the surface ECG, producing a
normal PR interval of 0.16 s. B. An atrial premature depolarization
(APD) blocks in the fast pathway. The impulse conducts over the slow pathway to
the His bundle and ventricles, producing a PR interval of 0.24 s. Because the
impulse is premature, conduction over the slow pathway occurs more slowly than
it would during sinus rhythm. C. A more premature atrial impulse blocks
in the fast pathway, conducting with increased delay in the slow pathway,
producing a PR interval of 0.28 s. The impulse conducts retrogradely up the
fast pathway producing a single atrial echo. Sustained reentry is prevented by
subsequent block in the slow pathway. D. A still more premature atrial
impulse blocks initially in the fast pathway, conducting over the slow pathway
with increasing delay producing a PR interval of 0.36 s. Retrograde conduction
occurs over the fast pathway and reentry occurs, producing a sustained
tachycardia (SVT).
Clinical Features
AV
nodal reentry may produce palpitations, syncope, and heart failure depending on
the rate and duration of the arrhythmia and the presence and severity of any
underlying heart disease. Hypotension and syncope may occur because of the
sudden loss of the atrial contribution to ventricular filling; this can also
lead to a marked increase in atrial pressure, acute pulmonary edema, and a
reduction in ventricular filling. Simultaneous atrial and ventricular
contraction produces cannon a waves with each heartbeat.
![]() Treatment
Treatment
In
patients without hypotension, vagal maneuvers, particularly carotid sinus
massage, can terminate the arrhythmia in 80% of cases. If hypotension is
present, raising the blood pressure by the cautious use of intravenous
phenylephrine in 0.1-mg increments may terminate the arrhythmia alone or in
combination with carotid sinus pressure. If these maneuvers are unsuccessful,
verapamil (2.5 to 10 mg intravenously) or adenosine (6 to 12 mg intravenously)
is the agent of choice. We prefer to use adenosine because of its extremely
short half-life, lessening the consequences of any side effects. Beta blockers
may also be used to slow or terminate the tachycardia but are agents of second
choice. Digitalis glycosides have a slower onset of action and should not
be used for acute therapy. When these drugs fail to terminate the tachycardia,
or when the tachycardia is recurrent, atrial or ventricular pacing via a
temporary pacemaker inserted pervenously may be used to terminate the
arrhythmia. However, if severe ischemia and/or hypotension is caused by the
tachycardia, dc cardioversion should be considered.
AV
nodal reentry can usually be prevented by the use of drugs that act primarily
on the antegrade slow pathway (such as digitalis, beta blockers, or calcium
channel antagonists) or on the fast pathway (class IA or IC; Table 230-4). We
favor initial therapy with beta blockers, calcium channel antagonists, or
digoxin because the risk-benefit ratio associated with treatment with these
agents is more favorable than that of IA or IC agents. Drugs most likely to
avert recurrences prevent induction of the arrhythmias by programmed
stimulation. This technique utilizes temporary pacemaker catheters connected to
a physiologic stimulator capable of variable rate pacing and stimulation with
one or more precisely timed premature impulses. In symptomatic patients who
require chronic therapy, radiofrequency catheter modification of the AV node
should be considered. This technique can cure AV nodal reentry in >90% of
cases and has been proven to be safe, although a 1 to 2% risk of AV block
requiring a permanent pacemaker exists.
AV
Reentrant Tachycardia
PSVT
due to AV reentry incorporates a concealed AV bypass tract as part of the
tachycardia circuit. Thus the impulse passes antegradely from the atria through
the AV node and His-Purkinje system to the ventricles and then retrogradely
through the (concealed) bypass tract back to the atrium. Patients with this
disorder manifest the same type of PSVT as do patients with the WPW syndrome
(see below), but the bypass tract cannot conduct in an antegrade direction
during sinus rhythm or other atrial tachyarrhythmias.
AV
reentrant tachycardia can be initiated and terminated by either APCs or VPCs.
Initiation of PSVT by a VPC is virtually diagnostic of AV reentry. Alternation
of the QRS complexes occurs in approximately one-third of such tachycardias.
Since atrial activation must follow ventricular activation during AV reentry,
the P wave usually occurs after the QRS complex (Fig. 230-7B).
Atrial
activation mapping is of major value in evaluating the origin of these
tachycardias. Most concealed bypass tracts are left-sided. Thus, during PSVT or
during ventricular pacing, the earliest activation sequence is recorded in the
left atrium, usually via a catheter in the coronary sinus. This eccentric
atrial activation is quite distinct from the normal retrograde activation
sequence in which the earliest activation of the atria is in the area of the AV
junction. The ability of a ventricular stimulus to conduct to the atrium at a
time when the bundle of His is refractory and the termination of the
tachycardia by a ventricular stimulus that does not reach the atrium are
diagnostic of retrograde conduction over a concealed bypass tract.
![]() Treatment
Treatment
This
is similar to the treatment for AV nodal reentry tachycardia. Although
pharmacologic agents may be used, patients who require chronic therapy should
be considered candidates for radiofrequency catheter ablation of the bypass
tract. This requires detailed electrophysiologic study to exclude other
arrhythmias that may be responsible for patients' symptoms and to determine the
location of the bypass tract(s). The efficacy of this procedure exceeds 90%,
with minimal risks. In the remaining small number of patients failing catheter
ablation, surgical ablation or pharmacologic therapy can be used.
Sinus Node Reentry and Other Atrial Tachycardias
Reentry
in the region of the sinus node or within the atria is invariably initiated by
APCs. These arrhythmias are less common than AV nodal or AV reentry and are
more often associated with underlying cardiac disease. During sinus node
reentry, the P-wave morphology is identical to that occurring in sinus rhythm,
but the PR interval is prolonged. This is in contrast to sinus tachycardia, in
which the PR interval tends to shorten. With intraatrial reentry, the P-wave
configuration differs from that during sinus rhythm, and the PR interval is
prolonged (Fig. 230-7C).
![]() Treatment
Treatment
Sinus
node and atrial reentrant arrhythmias are managed like other reentrant PSVTs,
except that catheter ablation is less successful because multiple foci may be
present.
Nonreentrant Atrial Tachycardias
These
may be a manifestation of digitalis intoxication or may be associated with
severe pulmonary or cardiac disease, with hypokalemia, or with the
administration of theophylline or adrenergic drugs. Multifocal atrial
tachycardia (MAT) (Fig. 230-9) is particularly common following theophylline
administration. By definition, MAT requires three or more consecutive P waves
of different morphologies at rates greater than 100 beats per minute. MAT
usually has an irregular ventricular rate because of varying AV conduction.
There is a high incidence of atrial fibrillation (50 to 70%) in patients with
MAT. Treatment should be directed at the underlying disorder. The
digitalis-induced arrhythmias are caused by triggered activity. In such atrial
tachycardias with AV block secondary to digitalis intoxication, the atrial rate
rarely exceeds 180 bpm, and typically 2:1 block is present. Atrial arrhythmias
precipitated by digitalis can usually be treated by withdrawal of the drug.
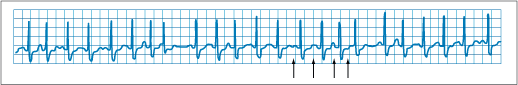
Figure 230-9: Multifocal atrial tachycardia. A lead I
rhythm strip demonstrates a multifocal atrial tachycardia defined by ![]() 3
consecutive P waves of variable morphology and rate >100 bpm (arrows).
3
consecutive P waves of variable morphology and rate >100 bpm (arrows).
Automatic
atrial tachycardias not caused by digitalis are difficult to terminate, and in
such cases the main goal of therapy should be to control the ventricular
response, either by drugs that affect the AV node, such as digitalis, beta
blockers, or calcium channel antagonists, or by ablation techniques. Catheter
ablation and surgery have been employed to eradicate the arrhythmia's focus or
create heart block for rate control.
Preexcitation (WPW)
Syndrome
The
most frequently encountered type of ventricular preexcitation is that
associated with AV bypass tracts (Fig. 230-20). These connections are composed
of strands of atrial-like muscle which may occur almost anywhere around the AV
rings. The term Wolff-Parkinson-White syndrome is applied to patients
with both preexcitation on the ECG and paroxysmal tachycardias. AV bypass
tracts can be associated with certain congenital abnormalities, the most
important of which is Ebstein's anomaly.
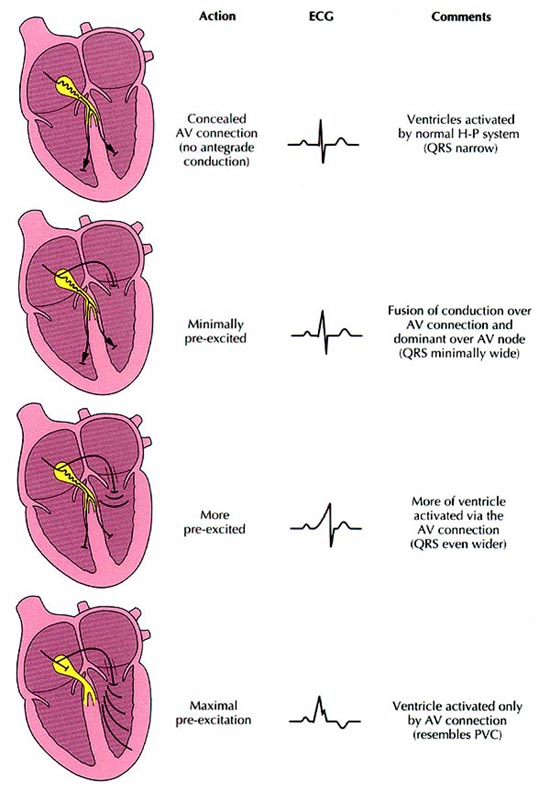
Figure 230-20: Conduction over an
accessory pathway. The morphology of a QRS complex in patients with an
accessory pathway (AP) is an example of fusion. If there is no antegrade
conduction over the AP (top) the AP may still conduct in the retrograde
direction but is considered concealed. When conduction over the AP is minimal
and atrioventricular (AV) node conduction predominates, the QRS becomes a
fusion of conduction over the A V nOde-His-Purkinje system and the AP. When
conduction to the ventricle over the AP predominates compared with AV nodal
conduction, the QRS represents a fusion of conduction over the AV node-HPS and
the AP, but the larger the portion of the ventricle activated by the AP, the
wider the QRS complex appears. H-P, His Purkinje; LA, left atrium; LV, left
ventricle; PVC, premature ventricular complex; RA, right atrium; RV, right
ventricle. [HR Grogin: Supraventricular tachycardia, in M Scheinman (volume
ed): Arrhythmias: Electrophysiologic Principles, in
AV
bypass tracts that conduct in an antegrade direction produce a typical ECG
pattern of a short PR interval (<0.12 s), a slurred upstroke of the QRS
complex (delta wave), and a wide QRS complex. This pattern results from a
fusion of activation of the ventricles over both the bypass tract and the AV
nodal His-Purkinje system (Fig. 230-10). The relative contribution of
activation over each system determines the amount of preexcitation.
Figure 230-10: ECG in Wolff-Parkinson-White
syndrome. There is a short PR interval (0.11 s), a wide QRS complex (0.12 s),
and slurring on the upstroke of the QRS produced by early ventricular
activation over the bypass tract (delta wave, d in lead I). The negative delta
waves in V1 are diagnostic of a right-sided bypass tract. Note the Q
wave (negative delta wave) in lead III, mimicking myocardial infarction.
During
PSVT in WPW, the impulse is usually conducted antegradely over the normal AV
system and retrogradely through the bypass tract. Rarely (approximately 5%),
tachycardias occurring in patients with WPW will exhibit a reverse pattern with
antegrade conduction through the bypass tract and retrograde conduction through
the normal AV system. This produces a tachycardia with a wide QRS complex in
which the ventricles are totally activated by the bypass tract. Atrial flutter
and AF also occur commonly in patients with WPW syndrome. Since the bypass
tract does not have the same decremental conducting properties as the AV node,
the ventricular responses during atrial flutter or fibrillation may be
unusually rapid and may cause ventricular fibrillation (VF).
The
goals of electrophysiologic evaluation in patients suspected of having the WPW
syndrome are (1) to confirm the diagnosis, (2) to localize the bypass tract and
determine how many bypass tracts are present, (3) to demonstrate the role of
the bypass tract in the genesis of the arrhythmias, (4) to determine the
potential for the development of possibly life-threatening rates during atrial flutter
or fibrillation, and (5) to evaluate therapeutic options.
![]() Treatment
Treatment
Pharmacologic
therapy is aimed at altering the electrophysiologic properties (i.e.,
refractoriness or conduction velocity) of one or more components of the
reentrant circuit. This is most often accomplished by agents such as beta
blockers or calcium channel blockers that slow conduction and increase
refractoriness of the AV node or by agents such as quinidine or flecainide that
slow conduction and increase refractoriness primarily in the bypass tract. Some
drugs may affect multiple sites (Fig. 230-11).
Figure 230-11: Site of action of
antiarrhythmic agents in the Wolff-Parkinson-White syndrome. The atrium,
ventricle, antegrade conduction through the AV node (![]() ),
retrograde conduction through the AV node (
),
retrograde conduction through the AV node (![]() ),
and the bypass tract are shown. Drugs on the left affect antegrade AV nodal
conduction.
),
and the bypass tract are shown. Drugs on the left affect antegrade AV nodal
conduction.
Acute
management of episodes of PSVT in patients with WPW syndrome is similar to that
of PSVT in patients with concealed bypass tracts.
In
patients with the WPW syndrome and AF, dc cardioversion should be carried out
if there is a life-threatening, rapid ventricular response. In
non-life-threatening situations, lidocaine (3 to 5 mg/kg) or procainamide (15
mg/kg) administered intravenously over 15 to 20 min will usually slow the
ventricular response. More recently, ibutilide has become available as an
alternative therapy for preexcitation tachycardia. Caution should be employed
when using digitalis or intravenous verapamil in patients with the WPW syndrome
and AF, since these drugs can shorten the refractory period of the accessory
pathway and can increase the ventricular rate, thereby placing the patient at
increased risk for VF. Chronic oral therapy with verapamil is not associated
with this risk. In addition to these drugs, beta-blocking agents are of no
utility in controlling the ventricular response during AF when conduction
proceeds over the bypass tract. Although atrial or ventricular pacing can
almost always terminate PSVT in patients with the WPW syndrome, they can induce
AF. As such, chronic pacemaker therapy is to be discouraged.
While
surgical ablation of bypass tracts offers a permanent cure of supraventricular
tachycardia (SVT) and most AFs associated with SVT, the advent of
radiofrequency catheter ablation has virtually eliminated the need for surgery.
Catheter ablation of bypass tracts is possible in >90% of patients and is
the treatment of choice in patients with symptomatic arrhythmias. It is safer,
more cost-effective, and just as successful as surgery. Nevertheless, surgical
ablation may be required in the occasional patient in whom catheter ablation
fails.
Nonparoxysmal Junctional Tachycardia
This
rhythm usually results from conditions that produce enhanced automaticity or
triggered activity in the AV junction and is most commonly due to digitalis
intoxication, inferior wall myocardial infarction, myocarditis, endogenous or
exogenous catecholamine excess, acute rheumatic fever, or valve surgery.
The
onset of nonparoxysmal junctional tachycardia is usually gradual, with a
"warm-up" period prior to stabilization of the rate, which can range
from 70 to 150 bpm, faster rates usually being associated with digitalis
intoxication. Nonparoxysmal junctional tachycardia is recognized by a QRS
complex identical to that of sinus rhythm. The rate can be influenced by
autonomic tone and can be increased by catecholamines, vagolytic agents, or
exercise and slowed somewhat by carotid sinus pressure. When this rhythm is due
to digitalis intoxication, it usually is associated with AV block and/or
dissociation. Soon after cardiac surgery, retrograde conduction is more likely
to be present because of the heightened sympathetic state.
![]() Treatment
Treatment
This
is directed toward elimination of the underlying etiologic factors. Since
digitalis is the most common cause of this rhythm, discontinuation of this drug
is indicated. If the rhythm is associated with other serious manifestations of
digitalis intoxication, such as ventricular or atrial irritability, active
intervention with lidocaine or a beta blocker may be useful, and in some
instances, use of digitalis antibodies (Fab fragments) should be considered.
Cardioversion of this rhythm should not be attempted, particularly in the
setting of digitalis intoxication. When AV conduction is intact, atrial pacing
can capture and override the junctional focus and provide the AV synchrony
necessary to maximize cardiac output. Nonparoxysmal junctional tachycardia is
usually not a chronic, recurrent problem, and attention to the acute
precipitating events can often resolve the tachycardia.
Ventricular Tachycardia
Sustained ventricular tachycardia is defined as VT that
persists for more than 30 s or requires termination because of hemodynamic
collapse. VT generally accompanies some form of structural heart disease, most
commonly chronic ischemic heart disease associated with a prior myocardial
infarction. Sustained VT may also be associated with nonischemic
cardiomyopathies, metabolic disorders, drug toxicity, or prolonged QT syndrome,
and it occurs occasionally in the absence of heart disease or other
predisposing factors.
The
ECG diagnosis of VT is suggested by a wide-complex QRS tachycardia at a rate
exceeding 100 bpm. The QRS configuration during any episode of VT may be
uniform (monomorphic), or it may vary from beat to beat (polymorphic). Bidirectional
tachycardia refers to VT that shows an alternation in QRS amplitude and
axis. Typically this appears as a QRS with a right bundle branch block pattern
with alternating superior (leftward) and inferior axes (rightward). While the
rhythm is usually quite regular, slight irregularity may exist. Atrial activity
may be dissociated from ventricular activity, or the atria may be depolarized
retrogradely. The onset of the tachycardia is generally abrupt, but in
nonparoxysmal tachycardias it can be gradual. Paroxysmal VT is usually
initiated by a VPC.
It
is important to distinguish SVT with aberration of intraventricular conduction
from VT because the clinical implications and management of these two
arrhythmias are totally different. The most important clinical predictor of VT
is the presence of structural heart disease. The observation of intermittent
cannon a waves and varying first heart sounds suggests AV dissociation
and is diagnostic of VT. In a majority of cases, the diagnosis can and should
be made by close examination of the 12-lead ECG. Pharmacologic maneuvers, such
as administration of intravenous verapamil or adenosine, can be hazardous and
should be avoided. It is always useful to have a 12-lead ECG recorded during
sinus rhythm for comparison with that during tachycardia. When the tracing
obtained during sinus rhythm demonstrates the same morphologic features as
those during the tachycardia, the diagnosis of PSVT with aberration is favored.
An infarction pattern on the sinus rhythm tracing suggests the potential
presence of the anatomic substrate necessary for VT. Characteristics of the
12-lead ECG during the tachycardia that suggest a ventricular origin for the
arrhythmia are (1) a QRS complex >0.14 s in the absence of antiarrhythmic
therapy, (2) AV dissociation (with or without fusion or captured beats) or
variable retrograde conduction (Fig. 230-12), (3) a superior QRS axis in the
presence of a right bundle branch block pattern, (4) concordance of the QRS
pattern in all precordial leads (i.e., all positive or all negative deflections),
and (5) other QRS patterns (morphology) with prolonged duration that are
inconsistent with typical right or left bundle branch block patterns. (See
Table 230-5 for a detailed synopsis of ECG criteria that favor the diagnosis of
VT over SVT for wide complex tachycardia.) A wide, complex, bizarre tachycardia
that is very irregular suggests AF with conduction over an AV bypass tract.
Similarly, a QRS complex in excess of 0.20 s is uncommon during VT in the
absence of drug therapy and is more common with preexcitation. Intravenous
verapamil will stop most recalcitrant SVTs involving the AV junction, but it is
rarely effective for VT. Because of this property, verapamil has been utilized
to attempt to differentiate SVT with aberrant conduction from VT. However, this
is extremely hazardous, since intravenous verapamil can precipitate cardiac
arrest in patients with VT.
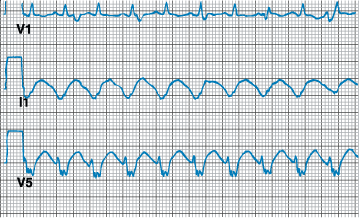
Figure 230-12: Ventricular tachycardia
with AV dissociation. P waves are dissociated from the underlying wide complex
rhythm (best seen on lead V1).
Table 230-5: Wide Complex Tachycardia
|
||||
|
||||
It
has been possible to replicate sustained uniform VT in more than 95% of
patients with this arrhythmia using programmed electrical stimulation. In most patients
the tachycardia is initiated with ventricular premature stimuli. A sustained
monomorphic VT with a morphology identical to that of the spontaneous
arrhythmia is the rule. The clinical significance of polymorphic VT initiated
by programmed stimulation is not clear, since more aggressive stimulation
(i.e., the use of three or four extrastimuli) can induce polymorphic VT and
even VF in some normal subjects and in patients who have never had a clinical
arrhythmia.
Sustained
uniform VT can be terminated by programmed stimulation or rapid pacing in at
least 75% of patients; the remainder require cardioversion. The ability to
reproducibly initiate and terminate a sustained, uniform VT permits assessment
of pharmacologic and electrical therapy of these arrhythmias.
The
reproducible termination of VT by programmed stimulation permits evaluation of
the effectiveness of antitachycardia pacemakers for long-term therapy of
paroxysmal episodes of arrhythmia. Unfortunately, rapid pacing, the most
effective form of therapy, can accelerate the tachycardia and/or produce VF.
Therefore, antitachycardia pacing is a viable form of therapy only when the
pacing device includes backup defibrillation capabilities.
Clinical Features
Symptoms
resulting from VT depend on the ventricular rate, duration of the tachycardia,
and presence and extent of underlying cardiac disease. When the tachycardia is
rapid and associated with severe myocardial dysfunction and cerebrovascular
disease, hypotension and syncope are common. However, the presence of
hemodynamic stability does not preclude a diagnosis of VT. The rate, loss of
the atrial contribution to ventricular filling, and abnormal sequence of
ventricular activation are important factors producing a decreased cardiac
output during VT.
The
prognosis of VT depends on the underlying disease state. If sustained VT
develops within the first 6 weeks following acute myocardial infarction, the
prognosis is poor, with a 75% mortality rate at 1 year. Patients with
nonsustained VT following myocardial infarction have a threefold greater risk
of death than a comparable group of patients without this arrhythmia. However,
a cause-and-effect relationship between the nonsustained tachycardia and
subsequent sudden death has not been established. Patients without heart
disease who have uniform VT have a good prognosis and an extremely low risk of
sudden death.
![]() Treatment
Treatment
The
risk-benefit ratio of treating each specific type of VT should be considered
before beginning therapy. This is important because antiarrhythmic agents can
produce or exacerbate the very arrhythmias that they are given to prevent. In
general, patients with VT but without organic heart disease have a benign
course; such patients with asymptomatic, nonsustained VT need not be treated
because their prognosis will not be affected. An exception is the patient with
congenital long QT syndrome. Such patients have recurrent polymorphic VT and a
high mortality from sudden death if untreated. Patients with sustained VT in
the absence of heart disease usually require therapy because the arrhythmia
causes symptoms. These tachycardias may respond to beta blockers; verapamil;
class IA, IC, or III agents (Fig. 230-21); or amiodarone. In patients with VT
and organic heart disease, if marked hemodynamic compromise is present or if
there is evidence of ischemia, congestive heart failure, or central nervous
system hypoperfusion, the rhythm should be promptly terminated by dc
cardioversion (see below). If the patient with organic heart disease tolerates
the VT well, pharmacologic therapy may be tried. Procainamide is probably the
most effective agent for acute therapy. It may or may not terminate the
tachycardia but almost always slows the rate. In stable patients in whom these
drugs do not terminate the arrhythmia, a pacing catheter can be inserted
pervenously into the right ventricular apex, and the tachycardia can be
terminated by overdrive pacing.
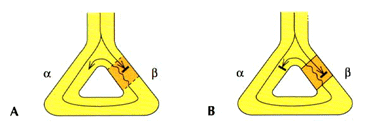
Figure 230-21: The effect of a drug
with class III antiarrhythmic action on a reentry circuit by sufficiently
prolonging the refractory period of path Ot, thus preventing reentry from
occurring (B).
Programmed
stimulation is probably the most effective way to select the appropriate
antiarrhythmic agent to prevent recurrent, sustained VT. After demonstrating
that the tachycardia can be initiated reproducibly in the absence of
antiarrhythmic agents, drugs can be studied serially, and the drug that
prevents initiation of the tachycardia can be selected; long-term (>2 years)
successful prevention of the arrhythmia can then be expected in 80% of patients
if a complete stimulation protocol is used following drug administration.
Failure to perform a complete protocol will lead to recurrences, which are
often blamed on the lack of utility of programmed stimulation as a method of
evaluating drug efficacy. Drug levels demonstrated to be successful in the
laboratory need to be maintained chronically. Unfortunately, prevention of
inducible VT is expected in only 50% of cases. Use of Holter monitor for guided
therapy, although advocated by some, is of less value.
Antitachycardia
pacing has been used as a means to terminate tachycardias that have been
reproducibly terminated by pacing in the electrophysiology laboratory.
Automatic antitachycardia pacing devices are not used alone because pacing
during VT may accelerate tachycardia, converting a stable arrhythmia into an
unstable one and resulting in severe hemodynamic compromise. However, devices
combining antitachycardia pacing with an ICD (see below) afford a "backup"
means of terminating unstable arrhythmias.
The
advent of endocardial catheter and intraoperative mapping led to the
development of surgical techniques for the management of VT. Activation mapping
permits localization of the site of origin of the arrhythmia. In centers in
which expertise in mapping is available, operation has been successfully
employed to cure tachycardias in the majority of patients in whom it has been
undertaken. Even though most patients with VT and ischemic heart disease have
markedly impaired left ventricular function and multivessel coronary artery
disease, the operative mortality rate has ranged between 8 and 15%. Following
operation, >90% of survivors are controlled either off (two-thirds of
patients) or on (one-third) antiarrhythmic agents that were previously
ineffective in controlling these rhythms. With the development of
radiofrequency ablation and refinement of mapping criteria to locate critical
sites of the VT circuit, precisely, catheter ablation can be performed as a
curative procedure in selected patients. In experienced centers cure of VT in
these selected patients approaches 75%.
Specific Types of VT
Torsade de pointes (Fig. 230-13) refers to VT characterized by
polymorphic QRS complexes that change in amplitude and cycle length, giving the
appearance of oscillations around the baseline. This rhythm is, by definition,
associated with QT prolongation. The latter may result from electrolyte
disturbances (particularly hypokalemia and hypomagnesemia), use of a variety of
antiarrhythmic drugs (especially quinidine), phenothiazines and tricyclic
antidepressants, liquid protein diets, intracranial events, and
bradyarrhythmias, particularly third-degree AV block. It also may occur as a
congenital anomaly that most often presents with torsade de pointes (syncope or
sudden death) at a young age.
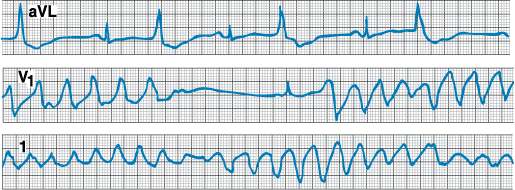
Figure 230-13: Rhythm strips of
patients with drug (disopyramide)-induced torsade de pointes. The polymorphic
ventricular tachycardia is associated with very long QT intervals.
The
electrocardiographic hallmark is polymorphic VT preceded by marked QT
prolongation, often in excess of 0.60 s. These patients often have multiple episodes
of nonsustained polymorphic VT associated with recurrent syncope, but they also
may develop VF and sudden cardiac death.
Therapy should be directed at removing the precipitating factors,
i.e., correcting metabolic abnormalities and removing drugs that have induced
the prolonged QT interval. In the setting of drug-induced torsade de pointes,
atrial or ventricular overdrive pacing and the administration of magnesium have
also been useful in terminating and preventing the arrhythmia. For patients with
the congenital prolonged QT interval syndrome, ![]() -adrenergic
blocking agents have been the mainstay of therapy; agents that shorten the QT
interval may also be useful (e.g., phenytoin). Cervicothoracic sympathectomy
has been proposed as a form of therapy for congenital prolonged QT syndrome,
but it is not often effective as the sole therapy. Pacing in combination with beta
blockers and sympathectomy has been used by some investigators when beta
blockers fail, but it is not uniformly successful and results in a Horner's
syndrome. More recently, ICDs with dual chambered pacing capability and beta
blockers have become the treatment of choice for patients with recurrent
episodes despite beta blockers.
-adrenergic
blocking agents have been the mainstay of therapy; agents that shorten the QT
interval may also be useful (e.g., phenytoin). Cervicothoracic sympathectomy
has been proposed as a form of therapy for congenital prolonged QT syndrome,
but it is not often effective as the sole therapy. Pacing in combination with beta
blockers and sympathectomy has been used by some investigators when beta
blockers fail, but it is not uniformly successful and results in a Horner's
syndrome. More recently, ICDs with dual chambered pacing capability and beta
blockers have become the treatment of choice for patients with recurrent
episodes despite beta blockers.
Polymorphic
tachycardias associated with normal QT intervals in patients with ischemic
heart disease that are initiated by "R-on-T" VPCs are probably caused
by reentry, and their treatment is totally different. This is not true torsade
de pointes. In such cases, class I or III agents may be the most effective form
of therapy and should be administered in full antiarrhythmic doses. However,
these arrhythmias may also result from acute, severe ischemia and will only
respond to abolition of the ischemia, usually by revascularization.
Accelerated idioventricular rhythm, also termed slow VT,
with a rate that ranges from 60 to 120 bpm, usually occurs in acute myocardial
infarction, often during reperfusion. It may also be seen following cardiac
operations; in patients with cardiomyopathy, rheumatic fever, or digitalis
intoxication; and in patients with no evidence of heart disease. The rhythm is
usually transient and rarely causes significant hemodynamic compromise or
symptoms.
Treatment
is rarely necessary and should usually be considered only if symptoms arise due
to impaired hemodynamics, most commonly due to AV dissociation. In most cases,
atropine can accelerate the sinus rate to overdrive the ventricular rhythm.
Ventricular Flutter and Ventricular Fibrillation
( Fig.
230-14;)
These
arrhythmias occur most often in patients with ischemic heart disease. They also
occur following administration of antiarrhythmic drugs, particularly those that
induce prolonged QT intervals and torsade de pointes (see above), in patients
with severe hypoxia or ischemia, and in those with WPW who develop AF with an
extremely rapid ventricular response (p. 1299). Electrical accidents frequently
cause cardiac arrest due to the development of VF. The onset of these
arrhythmias is rapidly followed by loss of consciousness and, if untreated,
death. Episodes of cardiac arrest recorded during Holter monitoring reveal that
approximately three-fourths of the sudden deaths are due to VT or VF.
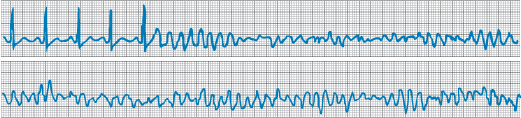
Figure 230-14: Ventricular
fibrillation. In a patient with coronary disease, ventricular fibrillation is
initiated by an early ventricular premature complex that produces a rapid
polymorphic ventricular tachycardia which rapidly degenerates to ventricular
fibrillation (note the undulating baseline with indistinguishable systole and
diastole).
In
patients with nonischemic VF, the onset usually begins with a short run of
rapid VT, which is initiated by a relatively late coupled VPC. In patients with
acute myocardial infarction or ischemia, however, VF is usually precipitated by
a single early ventricular complex beat falling on the T wave (the vulnerable
period), which produces a rapid VT that degenerates into VF (Fig. 230-14).
The
clinical setting in which VF occurs is important. Most patients who have
primary VF within the first 48 h of the onset of acute infarction have a good
long-term prognosis, with a very low rate of recurrence or sudden cardiac
death. Their short-term mortality may, however, be slightly increased. In contrast,
patients who experience VF unassociated with the development of acute
myocardial infarction have a recurrence rate of 20 to 30% in the year following
the event (Chap. 39).
Ventricular flutter usually appears as a sine wave with a rate
between 150 and 300 bpm. These oscillations make it impossible to assign a
specific morphology to the arrhythmia and in some cases to distinguish it from
rapid VT. VF is recognized by grossly irregular undulations of varying
amplitudes, contours, and rates (Fig. 230-14). Electrophysiologic studies have
demonstrated that regardless of the apparent gross irregularity on the surface
ECG, VF usually starts out with a rapid repetitive sequence of VT that
ultimately breaks down into multiple wavelets of reentry.
Electrophysiologic
studies have been useful in patients who have been resuscitated from cardiac
arrest. In approximately 70% of patients with prior infarction, programmed
stimulation can reproducibly initiate a sustained VT. Ablation may be possible
in some of these patients, particularly if the VT can be slowed so that it can
be mapped. Several recent secondary prevention trials have demonstrated
superior survival (3 years) in patients treated with ICDs versus amiodarone
(Table 230-6). However, in patients with ejection fractions >35% or <20%,
survival was comparable. Further subgroup analysis is necessary to identify
those patients most likely to be benefited by ICDs.
Table 230-6: Trials of ICD Therapy vs. Antiarrythmic Drugs (AAD): VT/VF Patients
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![]() Genetics
Genetics
Many
advances have been made in the identification of genes responsible for
syndromes associated with ventricular tachycardias and sudden cardiac death.
Four specific examples include the congenital long QT syndrome (LQTS),
hypertrophic obstructive cardiomyopathy (Chap. 238), arrhythmogenic right
ventricular dysplasia, and the Brugada syndrome. The latter is a recently
described disorder characterized by the electrocardiographic profile of a
pseudo bundle branch block pattern with ST elevation and terminal T-wave
inversion in leads V1-V3 (Fig. 230-15). The clinical
presentation is VF in patients with structurally normal hearts. A mutation in
the cardiac sodium channel, SCN 5A, is believed to be responsible. While the
same gene is responsible for the LQTS, the mutation is different in the two
syndromes.
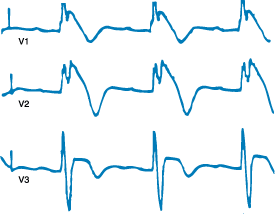
Figure 230-15: Brugada syndrome. Note
ST elevation with terminal T wave inversion in leads V1-V3
mimicking right bundle branch block.
![]() Treatment
Treatment
Pharmacologic Antiarrhythmic Therapy
Prior
to initiation of pharmacologic antiarrhythmic therapy, potential aggravating
factors such as transient metabolic abnormalities, congestive heart failure, or
acute ischemia must be corrected; in some cases this may suffice to control
arrhythmias. In addition, the potential role of drugs as a cause or
exacerbating factor in the development of the arrhythmia must be considered. It
must be recognized that we do not have a good understanding of the effects of
antiarrhythmic agents on the spontaneous onset of tachyarrhythmias. In some
cases, they may facilitate the onset.
Antiarrhythmic
drugs are used in three principal situations: (1) to terminate an acute
arrhythmia; (2) to prevent recurrence of an arrhythmia; and (3) to prevent a
life-threatening arrhythmia for which the patient is perceived to be at risk
but which has never occurred.
Most
currently available antiarrhythmic agents (Table 230-4) have a relatively low
toxic/therapeutic ratio; all can exert proarrhythmic effects (Table 230-7), and
therefore they may exacerbate underlying arrhythmias. Serum levels can be
determined for most currently available antiarrhythmic agents. Standards for
therapeutic and toxic levels can serve only as a rough guide for selecting the
appropriate dose in any individual patient. In the final analysis, the
therapeutic level in a given patient is the concentration that achieves the
desired antiarrhythmic effect, and the toxic level for each patient is the
concentration at which undesirable side effects occur. Since many adverse
effects are directly related to drug concentrations, the lowest serum level
that achieves an effective antiarrhythmic response should be chosen.
Table 230-7: Toxicity of Most Frequently Used
Antiarrhythmic Agents
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In
order to determine the therapeutic level for a patient, one must have a
standard to judge drug efficacy. For a patient with an incessant arrhythmia,
antiarrhythmic drugs may be administered empirically until the arrhythmia is
suppressed. If a reproducible precipitating factor such as exercise can be
identified, serial drug testing during such a provocative maneuver may be
performed. Unfortunately, most arrhythmias are sporadic and occur unpredictably
without identifiable precipitating factors. In these cases, if one waits to
observe spontaneous recurrences on each antiarrhythmic drug, assessment of drug
efficacy may require months. This type of assessment of efficacy may be
adequate for arrhythmias that are not life-threatening. However, this mode of
assessment is inadequate for arrhythmias that compromise hemodynamic stability,
result in syncope, or cause cardiac arrest. In such cases, two methods for
determination of arrhythmic drug efficacy have been utilized. The first, which
consists of continuous ECG monitoring in the control state and then in the
presence of antiarrhythmic drugs, has been used in order to determine the
effect that each drug has on spontaneous atrial or ventricular ectopy. This
method presupposes that the mechanism responsible for sustained arrhythmias is
the same as that causing isolated premature depolarizations (which may or may
not be true) and that therefore eradication of isolated ectopy will correlate
with prevention of sustained arrhythmias. This method has a number of limitations.
First, patients frequently show marked degrees of spontaneous variation in
frequency of ectopy, which may mimic antiarrhythmic drug effects. Second, 25 to
30% of patients with sustained ventricular arrhythmias such as VT or VF
demonstrate only rare spontaneous ectopy. Finally, many patients demonstrate a
dissociation between the effects of antiarrhythmic agents on spontaneous ectopy
and the effects of the same agent on sustained arrhythmias.
An
alternative method to assess drug efficacy is programmed stimulation. Numerous
studies have demonstrated that most clinically occurring supraventricular and
ventricular tachyarrhythmias may be reproducibly initiated and terminated
safely using this technique. Studies are performed initially in a baseline
state in the absence of antiarrhythmic drugs. If the patient's clinical
arrhythmia can be reproducibly initiated, then the ability of individual
antiarrhythmic drugs to prevent reinduction of the arrhythmia can be assessed
either after the drug is administered intravenously or after several days of
oral loading in order to achieve a steady-state serum concentration. Use of
this method assumes that (1) the induced and spontaneous arrhythmias are
identical, and (2) prevention of induction of arrhythmias will correlate with
prevention of recurrent spontaneous tachycardias on the same drug regimen. This
technique has been validated in patients with a variety of reentrant PSVTs, VT,
and VF. The technique is safe when carefully performed, the potential
complications being those of any intravascular catheterization. Appropriate
interpretation of the results of programmed stimulation is critically dependent
on correlating the patient's spontaneous arrhythmias with those induced in the
laboratory, with regard to rate and morphology, in order to be certain that the
arrhythmia induced in the laboratory represents the same arrhythmia that
occurred spontaneously and caused symptoms.
Classification
of Antiarrhythmic Drugs
A
number of classifications of antiarrhythmic drugs have been proposed; the most
frequently used is a modification of one proposed by Vaughan-Williams (Table
230-2). This classification is based in part on the ability of antiarrhythmic
drugs to modify the cardiac cellular (1) excitatory currents (Na+ or
Ca2+), (2) action potential duration, and (3) automaticity (phase 4
depolarization). These effects of the drugs on isolated cardiac cells are
thought to account for some of the antiarrhythmic properties of the drugs. Thus
depression of excitatory currents by class I and class IV antiarrhythmics
results in slowing of conduction velocity and may interrupt arrhythmias by
blocking conduction in areas of marginal excitability, where conduction
velocity is already slow. Class III antiarrhythmics allegedly exert their
action by increasing refractoriness through prolongation of the action
potential duration. However, this classification has a number of limitations.
The electrophysiologic effects of these drugs in vivo may differ from their
effects on isolated cells. Also, the effects of heart rate and fiber geometry
are not considered. Not all drugs (e.g., adenosine) fit into the
classifications. Finally, some drugs (e.g., amiodarone) exhibit properties
consistent with multiple classes. The uses, actions, and toxic actions of currently
available antiarrhythmic drugs are summarized in Tables 230-4 and 230-7.
Electrical
Therapy of Tachyarrhythmias
Pacemakers
Cardiac
pacing can be used to terminate and in selected cases prevent recurrent
supraventricular and ventricular arrhythmias. Because many tachyarrhythmias
appear to be due to a reentrant mechanism with the impulse traveling in a
circuit, a properly timed paced impulse can penetrate and prematurely
depolarize part of the circuit, rendering it refractory to the next circulating
wavefront and thereby interrupting the circus movement. Pacing therapy for
arrhythmias is generally reserved for patients whose arrhythmias are refractory
to drug therapy and who remain hemodynamically stable during the tachycardia.
All forms of pacing therapy require repeated demonstration of their
effectiveness and reliability in terminating the arrhythmias during
electrophysiologic testing prior to implantation of the pacing device.
The
type of pacing device and modality selected for arrhythmia termination depends
on (1) the rate of the tachycardia (rates >160 bpm are rarely terminated by
a single premature stimulus), (2) the type of arrhythmia (atrial flutter and VT
are rarely terminated by single extrastimuli), and (3) concomitant drug
therapy.
Because
many tachycardias cannot be terminated by single premature stimuli, pacemakers
have been developed that allow for multiple extrastimuli (burst pacing) to be
introduced. In the current era, antitachycardia pacing is almost exclusively
for ventricular arrhythmias because of the success of radiofrequency ablative
therapy for supraventricular arrhythmias.
Cardiac
pacing has also been used to prevent ventricular tachyarrhythmias. Polymorphic
VT associated with a long QT interval and bradycardia (torsade de pointes, p.
1304) is most likely to respond. Pacing the atrium and/or ventricle at rates
between 90 and 120 bpm appears to increase the homogeneity of electrical
recovery and markedly reduces the propensity for a recurrence of arrhythmias.
Pacemakers
may be self-contained or energized by an external radiofrequency source. The
self-contained pacemaker may function automatically [i.e., it incorporates an
arrhythmia recognition program (circuit)], or it may be activated by an
external magnet. The major advantage of a fully automatic system is that there
is no need for the patient to recognize the arrhythmia in order for termination
to occur. The advantages of the externally activated system (rarely used today)
include (1) the decreased risk of unnecessary treatment because of faulty
sensing, and (2) the opportunity to initiate monitoring at the time of
attempted termination of arrhythmia. This type of monitoring is frequently
helpful if pacing techniques are employed to terminate VT, given the risk of
acceleration of the arrhythmia by pacing.
The
limitations of pacing therapy are primarily related to (1) the changes in the
characteristics of the arrhythmia over time such that programmed pacing
parameters no longer terminate the tachycardia, (2) the risk of acceleration of
the tachycardia with the development of AF when stimulating the atrium and the
development of rapid VT and VF when stimulating the ventricles, and (3)
inappropriate recognition of supraventricular tachyarrhythmias as ventricular
tachycardias, leading to delivery of therapy unnecessarily, which can initiate
VT or VF. Future pacing generators that can perform cardioversion and
defibrillation will increase the applicability of pacing therapy for the
treatment of arrhythmias (see below).
Cardioversion
and Defibrillation
Electrical
cardioversion and defibrillation remain the most reliable methods for
terminating arrhythmias. By depolarizing all or at least a large portion of
excitable myocardium in a near homogeneous fashion, the electrical shock can
interrupt reentrant arrhythmias. External cardioversion is routinely performed
by placing two paddles 12 cm in diameter in firm contact with the chest wall,
with one paddle usually located to the right of the sternum at the level of the
second rib and the other in the left anterior axillary line in the fifth
intercostal space. If the patient is conscious, a short-acting barbiturate to
act as an anesthetic or an amnesic drug such as diazepam or medazolam should be
administered to prevent patient discomfort. A person skilled in maintaining an
airway should be present.
Energy
is delivered synchronously with the QRS complex for all arrhythmias except
ventricular flutter and VF, since asynchronous shocks can produce VF. The
amount of energy used will vary with the type of tachycardia being treated.
With the exception of AF, SVTs can frequently be terminated with energy levels
in the range of 25 to 50 W · s, while AF usually requires ![]() 100
W · s for termination. For terminating VT, energy levels
100
W · s for termination. For terminating VT, energy levels ![]() 100
W · s should probably be employed. While energies as low as 25 W · s may be
used successfully, they also have a higher incidence of producing VF or AF. At
least 200 W · s of energy should be used for initial attempts at terminating
VF. If the initial shock fails, all repeated attempts at defibrillation should
be with the maximum energy that the defibrillator is capable of delivering (320
to 400 W · s).
100
W · s should probably be employed. While energies as low as 25 W · s may be
used successfully, they also have a higher incidence of producing VF or AF. At
least 200 W · s of energy should be used for initial attempts at terminating
VF. If the initial shock fails, all repeated attempts at defibrillation should
be with the maximum energy that the defibrillator is capable of delivering (320
to 400 W · s).
Indications
for cardioversion depend on the clinical setting and the patient's general
condition. Any tachycardia (except sinus tachycardia) that produces
hypotension, myocardial ischemia, or heart failure warrants consideration of
prompt termination using external cardioversion. Arrhythmias that fail to
terminate with pharmacologic therapy may also be terminated by electrical
cardioversion. Transient bradycardias and supraventricular and ventricular
irritability following cardioversion are common and usually do not warrant
antiarrhythmic intervention.
Implanted
Cardioverter/Defibrillator (Fig. 230-22)
ICD
devices have been developed that will promptly recognize and terminate
life-threatening ventricular arrhythmias. These devices can deliver <1 to 40
W · s, the amount of which can be programmed. Current devices have
antitachycardia pacing capabilities such that VT can be sensed and terminated
without resorting to a painful shock. In such devices, high-energy shocks are
reserved for hypotensive VT, acceleration of VT, or failure to terminate VT
after a programmed duration (Fig. 230-16). ICDs now can be implanted
transvenously, and some are small enough to be implanted in a manner similar to
pacemakers. Clinical trials testing the function of these devices in patients
with drug-refractory ventricular arrhythmias have demonstrated survival from
sudden death at 1 year ranging between 92 and 100%. Currently, ICDs should be
considered for patients with VT that is not hemodynamically tolerated. As
mentioned earlier, recent randomized trials suggest that ICDs confer improved
mortality over amiodarone in patients with hemodynamically untolerated VT and a
cardiac arrest not due to reversible causes (Table 230-6). Finally, they are
indicated for patients with depressed left ventricular function, prior
myocardial infarction, nonsustained and sustained VT at electrophysiologic
study (Table 230-8). Guidelines for their use are given in Table 230-9.
Figure
230-16: Normally
functioning implantable defibrillators. A continuous Holter monitoring tracing
is shown. On the top strip, a rapid polymorphic tachycardia is initiated which
beats more uniformly. The automatic implantable cardioverter/defibrillator
(ICD) senses the rhythm and delivers a shock which restores sinus rhythm.
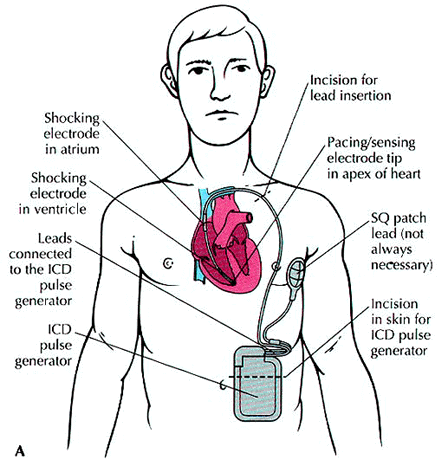
Figure
230-22: Nonthoracotomy
lead system for implantable defibrillator. [JL Cockrell, A Siu: Implantable
defibrillators for the management of cardiac rhythm disorders, in M Scheinman
(volume ed): Arrhythmias: Electrophysiologic Principles, in
Table
230-8: ICD Therapy for Primary
Prevention of Sudden Cardiac Death
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table
230-9: ACC/AHA Guidelines for ICD Implantation
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||
The
most frequent problem with the ICD has been its inappropriate discharge in the
absence of sustained ventricular arrhythmias. Additional potential problems include
an increase in defibrillation threshold and decrease in tachycardia rates below
the rate cut-off of the device in response to many antiarrhythmic drugs.
Permanently implanted ventricular pacemakers may interfere with the device's
ability to sense VF. This can be avoided by using committed bipolar pacing
systems that are better able to sense local ventricular activity. Diagnostic
features of newer, all-in-one devices are able to identify the probable cause
of an ICD discharge (e.g., AF, SVT, fractured lead) and to adjust pharmacologic
therapy or reprogram the device to avoid such inappropriate shocks. These newer
devices have the capability to take a "second look" prior to shock
delivery and thus may abort delivery for self-terminating arrhythmias. In
addition, the range of candidates suitable for implantation will be expanded
because the newer devices have the capability of shock therapy for patients
whose arrhythmias do not cause loss of consciousness.
Newer
generations of ICDs are smaller and frequently allow placement of a second lead
in the right atria. This lead senses atrial activity and provides enhanced
discrimination of atrial from ventricular electrical activity. This enhanced
discrimination of SVT from VT prevents inappropriate shocks for SVT that may be
misinterpreted as VT and allows the device to switch from a dual-chamber to a
single-chamber device should an SVT-like AF develop. These dual-chamber devices
also allow AV sequential pacing. Finally, ICDs are now available that have
defibrillation coils in the right atrium as well as right ventricle. These
devices are suited for patients with infrequent but highly symptomatic atrial
fibrillation. Patients with these atrial defibrillators can activate the device
themselves and terminate their atrial fibrillation without going to the
hospital.
Ablative Therapy for Arrhythmias
Catheter-based
mapping techniques have provided a nonoperative approach to the identification
and cure of a variety of arrhythmias. In fact, catheter ablation techniques are
now the procedures of choice for symptomatic patients with (1) concealed or
manifest (WPW) bypass tracts, (2) AV nodal reentrant SVT, (3) typical atrial
flutter, and (4) poorly controlled ventricular responses to atrial arrhythmias,
most commonly AF. Successful ablation of bypass tracts and modifications of the
AV node by radiofrequency energy are extremely successful and cost-effective
and are the procedure of choice for patients with recurrent episodes. The
creation of AV block with implantation of a pacemaker is the method of choice
in managing patients with AF and poorly controlled ventricular response.
Idiopathic VTs (Fig. 230-17) and some VTs that are associated with coronary
artery disease are also amenable to ablation, but the result is less successful
than for ablation of SVTs.
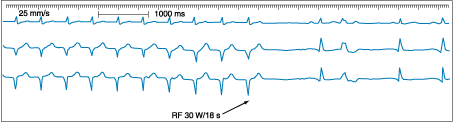
Figure 230-17: Radiofrequency ablation
of idiopathic ventricular tachycardia in a patient with prior infarction. Leads
I, II, and III are shown. Ventricular tachycardia with a right bundle branch
block and left axis deviation is shown in a patient with a prior inferior wall
infarction. Radiofrequency applied to the site of origin terminates the
tachycardia.
Surgical
therapy is now relegated to cases of sustained VT associated with coronary
artery disease when operative intervention is needed for coronary bypass
surgery and/or aneurysmectomy or VT associated with specific structural
abnormalities (e.g., idiopathic left ventricle aneurysm, s/p surgery for
tetralogy of Fallot). It also may be undertaken for the unusual instances of
failed catheter ablation for SVTs associated with bypass tracts.