Peptic Ulcer Disease
Burning
epigastric pain exacerbated by fasting and improved with meals is a symptom
complex associated with peptic ulcer disease (PUD). An ulcer is defined
as disruption of the mucosal integrity of the stomach and/or duodenum leading to
a local defect or excavation due to active inflammation. Ulcers occur within
the stomach and/or duodenum and are often chronic in nature. Acid peptic
disorders are very common in the United States, with 4 million individuals (new
cases and recurrences) affected per year. Lifetime prevalence of PUD in the
United States is approximately 12% in men and 10% in women. Moreover, an
estimated 15,000 deaths per year occur as a consequence of complicated PUD. The
financial impact of these common disorders has been substantial, with an
estimated burden on health care costs of >$15 billion per year in the United
States.
Gastric Physiology
Despite
the constant attack on the gastroduodenal mucosa by a host of noxious agents
(acid, pepsin, bile acids, pancreatic enzymes, drugs, and bacteria), integrity
is maintained by an intricate system that provides mucosal defense and repair.
Gastric Anatomy
The
gastric epithelial lining consists of rugae that contain microscopic gastric
pits, each branching into four or five gastric glands made up of highly
specialized epithelial cells. The makeup of gastric glands varies with their
anatomic location. Glands within the gastric cardia comprise <5% of the
gastric gland area and contain mucous and endocrine cells. The majority of gastric
glands (75%) are found within the oxyntic mucosa and contain mucous neck,
parietal, chief, endocrine, and enterochromaffin cells (Fig. 285-1). Pyloric
glands contain mucous and endocrine cells (including gastrin cells) and are
found in the antrum.
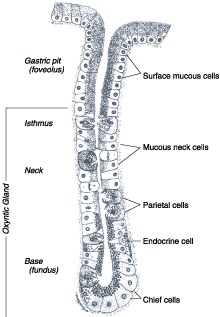
Figure 285-1: Diagramatic representation of the oxyntic
gastric gland.
The
parietal cell, also known as the oxyntic cell, is usually found in the neck, or
isthmus, or the oxyntic gland. The resting, or unstimulated, parietal cell has
prominent cytoplasmic tubulovesicles and intracellular canaliculi containing
short microvilli along its apical surface (Fig. 285-2). H+, K+-ATPase
is expressed in the tubulovesicle membrane; upon cell stimulation, this
membrane, along with apical membranes, transforms into a dense network of
apical intracellular canaliculi containing long microvilli. Acid secretion, a
process requiring high energy, occurs at the apical canalicular surface.
Numerous mitochondria (30 to 40% of total cell volume) generate the energy
required for secretion.
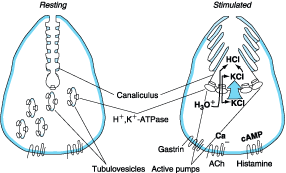
Figure 285-2: Gastric parietal cell undergoing
transformation after secretagogue-mediated stimulation.
Gastroduodenal Mucosal Defense
The
gastric epithelium is under a constant assault by a series of endogenous
noxious factors including HCl, pepsinogen/pepsin, and bile salts. In addition,
a steady flow of exogenous substances such as medications, alcohol, and
bacteria encounter the gastric mucosa. A highly intricate biologic system is in
place to provide defense from mucosal injury and to repair any injury that may
occur.
The
mucosal defense system can be envisioned as a three-level barrier, composed of
preepithelial, epithelial, and subepithelial elements (Fig. 285-3). The first
line of defense is a mucus-bicarbonate layer, which serves as a physicochemical
barrier to multiple molecules including hydrogen ions. Mucus is secreted in a
regulated fashion by gastroduodenal surface epithelial cells. It consists
primarily of water (95%) and a mixture of lipids and glycoproteins. Mucin is
the constituent glycoprotein that, in combination with phospholipids (also
secreted by gastric mucous cells), forms a hydrophobic surface with fatty acids
that extend into the lumen from the cell membrane. The mucous gel functions as
a nonstirred water layer impeding diffusion of ions and molecules such as
pepsin. Bicarbonate, secreted by surface epithelial cells of the gastroduodenal
mucosa into the mucous gel, forms a pH gradient ranging from 1 to 2 at the
gastric luminal surface and reaching 6 to 7 along the epithelial cell surface.
Bicarbonate secretion is stimulated by calcium, prostaglandins, cholinergic
input, and luminal acidification.
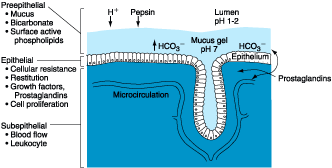
Figure 285-3: Components involved in providing
gastroduodenal mucosal defense and repair.
Surface
epithelial cells provide the next line of defense through several factors,
including mucus production, epithelial cell ionic transporters that maintain
intracellular pH and bicarbonate production, and intracellular tight junctions.
If the preepithelial barrier were breached, gastric epithelial cells bordering
a site of injury can migrate to restore a damaged region (restitution).
This process occurs independent of cell division and requires uninterrupted
blood flow and an alkaline pH in the surrounding environment. Several growth
factors including epidermal growth factor (EGF), transforming growth factor
(TGF) ![]() ,
and basic fibroblast growth factor (FGF) modulate the process of restitution.
Larger defects that are not effectively repaired by restitution require cell
proliferation. Epithelial cell regeneration is regulated by prostaglandins and
growth factors such as EGF and TGF-
,
and basic fibroblast growth factor (FGF) modulate the process of restitution.
Larger defects that are not effectively repaired by restitution require cell
proliferation. Epithelial cell regeneration is regulated by prostaglandins and
growth factors such as EGF and TGF-![]() .
In tandem with epithelial cell renewal, formation of new vessels (angiogenesis)
within the injured microvascular bed occurs. Both FGF and vascular endothelial
growth factor (VEGF) are important in regulating angiogenesis in the gastric
mucosa.
.
In tandem with epithelial cell renewal, formation of new vessels (angiogenesis)
within the injured microvascular bed occurs. Both FGF and vascular endothelial
growth factor (VEGF) are important in regulating angiogenesis in the gastric
mucosa.
An
elaborate microvascular system within the gastric submucosal layer is the key
component of the subepithelial defense/repair system. A rich submucosal
circulatory bed provides HCO3-, which neutralizes the
acid generated by parietal cell secretion of HCl. Moreover, this
microcirculatory bed provides an adequate supply of micronutrients and oxygen
while removing toxic metabolic by-products.
Prostaglandins
play a central role in gastric epithelial defense/repair (Fig. 285-4). The
gastric mucosa contains abundant levels of prostaglandins. These metabolites of
arachidonic acid regulate the release of mucosal bicarbonate and mucus, inhibit
parietal cell secretion, and are important in maintaining mucosal blood flow
and epithelial cell restitution. Prostaglandins are derived from esterified
arachidonic acid, which is formed from phospholipids (cell membrane) by the
action of phospholipase A2. A key enzyme that controls the
rate-limiting step in prostaglandin synthesis is cyclooxygenase (COX), which is
present in two isoforms (COX-1, COX-2), each having distinct characteristics
regarding structure, tissue distribution, and expression. COX-1 is expressed in
a host of tissues including the stomach, platelets, kidneys, and endothelial
cells. This isoform is expressed in a constitutive manner and plays an important
role in maintaining the integrity of renal function, platelet aggregation, and
gastrointestinal mucosal integrity. In contrast, the expression of COX-2 is
inducible by inflammatory stimuli, and it is expressed in macrophages,
leukocytes, fibroblasts, and synovial cells. The beneficial effects of
nonsteroidal anti-inflammatory drugs (NSAIDs) on tissue inflammation are due to
inhibition of COX-2; the toxicity of these drugs (e.g., gastrointestinal
mucosal ulceration and renal dysfunction) is related to inhibition of the COX-1
isoform. The highly COX-2-selective NSAIDs have the potential to provide the
beneficial effect of decreasing tissue inflammation while minimizing toxicity
in the gastrointestinal tract (see below).
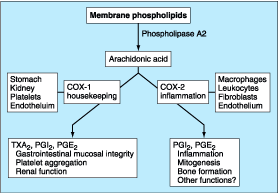
Figure 285-4: Schematic representation of the steps
involved in synthesis of prostaglandin E2 (PGE2) and
prostacyclin (PGI2). Characteristics and distribution of the cyclooxgenase
(COX) enzymes 1 and 2 are also shown. TXA2, thromboxane A2.
Physiology of Gastric Secretion
Hydrochloric
acid and pepsinogen are the two principal gastric secretory products capable of
inducing mucosal injury. Acid secretion should be viewed as occurring under
basal and stimulated conditions. Basal acid production occurs in a circadian
pattern, with highest levels occurring during the night and lowest levels
during the morning hours. Cholinergic input via the vagus nerve and
histaminergic input from local gastric sources (see below) are the principal
contributors to basal acid secretion. Stimulated gastric acid secretion occurs
primarily in three phases based on the site where the signal originates
(cephalic, gastric, and intestinal). Sight, smell, and taste of food are the
components of the cephalic phase, which stimulates gastric secretion via the
vagus nerve. The gastric phase is activated once food enters the stomach. This
component of secretion is driven by nutrients (amino acids and amines) that
directly stimulate the G cell to release gastrin, which in turn activates the
parietal cell via direct and indirect mechanisms (see below). Distention of the
stomach wall also leads to gastrin release and acid production. The last phase
of gastric acid secretion is initiated as food enters the intestine and is
mediated by luminal distention and nutrient assimilation. A series of pathways
that inhibit gastric acid production are also set into motion during these
phases. The gastrointestinal hormone somatostatin is released from endocrine
cells found in the gastric mucosa (D cells) in response to HCl. Somatostatin
can inhibit acid production by both direct (parietal cell) and indirect
mechanisms [decreased histamine release from enterochromaffin-like (ECL) cells
and gastrin release from G cells]. Additional neural (central and peripheral)
and hormonal (secretin, cholecystokinin) factors play a role in
counterbalancing acid secretion. Under physiologic circumstances, these phases
are occurring simultaneously.
The
acid-secreting parietal cell is located in the oxyntic gland, adjacent to other
cellular elements (ECL cell, D cell) important in the gastric secretory process
(Fig. 285-5). This unique cell also secretes intrinsic factor. The parietal
cell expresses receptors for several stimulants of acid secretion including
histamine (H2), gastrin (cholecystokinin B/gastrin receptor) and
acetylcholine (muscarinic, M3). Each of these are G protein-linked,
seven transmembrane-spanning receptors. Binding of histamine to the H2
receptor leads to activation of adenylate cyclase and an increase in cyclic
AMP. Activation of the gastrin and muscarinic receptors results in activation
of the protein kinase C/phosphoinositide signaling pathway. Each of these
signaling pathways in turn regulates a series of downstream kinase cascades,
which control the acid-secreting pump, H+, K+-ATPase. The
discovery that different ligands and their corresponding receptors lead to
activation of different signaling pathways explains the potentiation of acid
secretion that occurs when histamine and gastrin or acetylcholine are combined.
More importantly, this observation explains why blocking one receptor type (H2)
decreases acid secretion stimulated by agents that activate a different pathway
(gastrin, acetylcholine). Parietal cells also express receptors for ligands
that inhibit acid production (prostaglandins, somatostatin, and EGF).
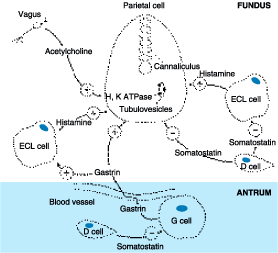
Figure 285-5: Regulation of gastric acid secretion at
the cellular level. ECL cell, enterochromaffin-like cell.
The
enzyme H+, K+-ATPase is responsible for generating the
large concentration of H+. It is a membrane-bound protein that
consists of two subunits, ![]() and
and
![]() .
The active catalytic site is found within the
.
The active catalytic site is found within the ![]() subunit;
the function of the
subunit;
the function of the ![]() subunit
is unclear. This enzyme uses the chemical energy of ATP to transfer H+
ions from parietal cell cytoplasm to the secretory canaliculi in exchange for K+.
The H+,K+-ATPase is located within the secretory
canaliculus and in nonsecretory cytoplasmic tubulovesicles. The tubulovesicles
are impermeable to K+, which leads to an inactive pump in this
location. The distribution of pumps between the nonsecretory vesicles and the
secretory canaliculus varies according to parietal cell activity (Fig. 285-2).
Under resting conditions, only 5% of pumps are within the secretory
canaliculus, whereas upon parietal cell stimulation, tubulovesicles are
immediately transferred to the secretory canalicular membrane, where 60 to 70%
of the pumps are activated. Proton pumps are recycled back to the inactive
state in cytoplasmic vesicles once parietal cell activation ceases.
subunit
is unclear. This enzyme uses the chemical energy of ATP to transfer H+
ions from parietal cell cytoplasm to the secretory canaliculi in exchange for K+.
The H+,K+-ATPase is located within the secretory
canaliculus and in nonsecretory cytoplasmic tubulovesicles. The tubulovesicles
are impermeable to K+, which leads to an inactive pump in this
location. The distribution of pumps between the nonsecretory vesicles and the
secretory canaliculus varies according to parietal cell activity (Fig. 285-2).
Under resting conditions, only 5% of pumps are within the secretory
canaliculus, whereas upon parietal cell stimulation, tubulovesicles are
immediately transferred to the secretory canalicular membrane, where 60 to 70%
of the pumps are activated. Proton pumps are recycled back to the inactive
state in cytoplasmic vesicles once parietal cell activation ceases.
The
chief cell, found primarily in the gastric fundus, synthesizes and secretes
pepsinogen, the inactive precursor of the proteolytic enzyme pepsin. The acid
environment within the stomach leads to cleavage of the inactive precursor to
pepsin and provides the low pH (<2.0) required for pepsin activity. Pepsin
activity is significantly diminished at a pH of 4 and irreversibly inactivated
and denatured at a pH of ![]() 7.
Many of the secretagogues that stimulate acid secretion also stimulate
pepsinogen release. The precise role of pepsin in the pathogenesis of PUD
remains to be established.
7.
Many of the secretagogues that stimulate acid secretion also stimulate
pepsinogen release. The precise role of pepsin in the pathogenesis of PUD
remains to be established.
Pathophysiologic Basis of Peptic Ulcer Disease
PUD
encompasses both gastric and duodenal ulcers. Ulcers are defined as a break in
the mucosal surface >5 mm in size, with depth to the submucosa. Duodenal
(DU) and gastric ulcers (GU) share many common features in terms of
pathogenesis, diagnosis, and treatment, but several factors distinguish them
from one another.
Epidemiology
Duodenal Ulcers
DUs
are estimated to occur in 6 to 15% of the western population. The incidence of
DUs declined steadily from 1960 to 1980 and has remained stable since then. The
death rates, need for surgery, and physician visits have decreased by >50%
over the past 30 years. The reason for the reduction in the frequency of DUs is
likely related to the decreasing frequency of Helicobacter pylori.
Before the discovery of H. pylori, the natural history of DUs was
typified by frequent recurrences after initial therapy. Eradication of H.
pylori has greatly reduced these recurrence rates.
Gastric Ulcers
GUs
tend to occur later in life than duodenal lesions, with a peak incidence
reported in the sixth decade. More than half of GUs occur in males and are less
common than DUs, perhaps due to the higher likelihood of GUs being silent and
presenting only after a complication develops. Autopsy studies suggest a
similar incidence of DUs and GUs.
Pathology
Duodenal Ulcers
DUs
occur most often in the first portion of duodenum (>95%), with ~90% located
within 3 cm of the pylorus. They are usually ![]() 1
cm in diameter but can occasionally reach 3 to 6 cm (giant ulcer). Ulcers are
sharply demarcated, with depth at times reaching the muscularis propria. The
base of the ulcer often consists of a zone of eosinophilic necrosis with
surrounding fibrosis. Malignant duodenal ulcers are extremely rare.
1
cm in diameter but can occasionally reach 3 to 6 cm (giant ulcer). Ulcers are
sharply demarcated, with depth at times reaching the muscularis propria. The
base of the ulcer often consists of a zone of eosinophilic necrosis with
surrounding fibrosis. Malignant duodenal ulcers are extremely rare.
Gastric Ulcers
In
contrast to DUs, GUs can represent a malignancy. Benign GUs are most often
found distal to the junction between the antrum and the acid secretory mucosa.
This junction is variable, but in general the antral mucosa extends about two
thirds of the distance of the lesser curvature and one third the way up the
greater curvature. Benign GUs are quite rare in the gastric fundus and are
histologically similar to DUs. Benign GUs associated with H. pylori are
associated with antral gastritis. In contrast, NSAID-related GUs are not
accompanied by chronic active gastritis but may instead have evidence of a
chemical gastropathy.
Pathophysiology
It
is now clear that H. pylori and NSAID-induced injury account for the
majority of DUs. Gastric acid contributes to mucosal injury but does not play a
primary role.
Duodenal Ulcers
Many
acid secretory abnormalities have been described in DU patients (Table 285-1).
Of these, average basal and nocturnal gastric acid secretion appear to be
increased in DU patients as compared to control; however, the level of overlap
between DU patients and control subjects is substantial. The reason for this
altered secretory process is unclear, but H. pylori infection may
contribute to this finding. Accelerated gastric emptying of liquids has been
noted in some DU patients but is not consistently observed; its role in DU
formation, if any, is unclear. Bicarbonate secretion is significantly decreased
in the duodenal bulb of patients with an active DU as compared to control
subjects. H. pylori infection may also play a role in this process.
Table 285-1: Reported Pathophysiologic
Abnormalities in Patients with Duodenal Ulcers
|
|||||||||||||||||||||||||||
Gastric Ulcer
As
in DUs, the majority of GUs can be attributed to either H. pylori or
NSAID-induced mucosal damage. GUs that occur in the prepyloric area or those in
the body associated with a DU or a duodenal scar are similar in pathogenesis to
DUs. Gastric acid output (basal and stimulated) tends to be normal or decreased
in GU patients. When GUs develop in the presence of minimal acid levels,
impairment of mucosal defense factors may be present.
Abnormalities
in resting and stimulated pyloric sphincter pressure with a concomitant
increase in duodenal gastric reflux have been implicated in some GU patients.
Although bile acids, lysolecithin, and pancreatic enzymes may injure gastric
mucosa, a definite role for these in GU pathogenesis has not been established.
Delayed gastric emptying of solids has been described in GU patients but has
not been reported consistently. The observation that patients who have
undergone disruption of the normal pyloric barrier (pyloroplasty,
gastroenterostomy) often have superficial gastritis without frank ulceration
decreases enthusiasm for duodenal gastric reflux as an explanation for GU
pathogenesis.
H. pylori and acid peptic disorders
Gastric
infection with the bacterium H. pylori accounts for the majority of PUD.
This organism also plays a role in the development of gastric
mucosal-associated lymphoid tissue (MALT) lymphoma and gastric adenocarcinoma.
Although the entire genome of H. pylori has been sequenced, it is still
not clear how this organism, which is in the stomach, causes ulceration in the
duodenum, or whether its eradication will lead to a decrease in gastric cancer.
The Bacterium
The
bacterium, initially named Campylobacter pyloridis, is a gram-negative
microaerophilic rod found most commonly in the deeper portions of the mucous
gel coating the gastric mucosa or between the mucous layer and the gastric
epithelium. It may attach to gastric epithelium but under normal circumstances
does not appear to invade cells. It is strategically designed to live within
the aggressive environment of the stomach. It is S-shaped (~0.5 × 3 ![]() m
in size) and contains multiple sheathed flagella. Initially, H. pylori
resides in the antrum but, over time, migrates towards the more proximal
segments of the stomach. The organism is capable of transforming into a coccoid
form, which represents a dormant state that may facilitate survival in adverse
conditions. The bacterium expresses a host of factors that contribute to its
ability to colonize the gastric mucosa and produce mucosal injury. Several of
the key bacterial factors include urease (converting urea to NH3 and
water, thus alkalinizing the surrounding acidic environment), catalase, lipase,
adhesins, platelet-activating factor, cytotoxin-associated gene protein (Cag
A), pic B (induces cytokines), and vacuolating cytotoxin (Vac A). Multiple
strains of H. pylori exist and are characterized by their ability to
express several of these factors (Cag A, Vac A, etc.). It is possible that the
different diseases related to H. pylori infection can be attributed to
different strains of the organism with distinct pathogenic features.
m
in size) and contains multiple sheathed flagella. Initially, H. pylori
resides in the antrum but, over time, migrates towards the more proximal
segments of the stomach. The organism is capable of transforming into a coccoid
form, which represents a dormant state that may facilitate survival in adverse
conditions. The bacterium expresses a host of factors that contribute to its
ability to colonize the gastric mucosa and produce mucosal injury. Several of
the key bacterial factors include urease (converting urea to NH3 and
water, thus alkalinizing the surrounding acidic environment), catalase, lipase,
adhesins, platelet-activating factor, cytotoxin-associated gene protein (Cag
A), pic B (induces cytokines), and vacuolating cytotoxin (Vac A). Multiple
strains of H. pylori exist and are characterized by their ability to
express several of these factors (Cag A, Vac A, etc.). It is possible that the
different diseases related to H. pylori infection can be attributed to
different strains of the organism with distinct pathogenic features.
Epidemiology
The
prevalence of H. pylori varies throughout the world and depends to a
great extent on the overall standard of living in the region. In developing
parts of the world, 80% of the population may be infected by the age of 20. In
contrast, in the United States, this organism is rare in childhood. The overall
prevalence of H. pylori in the United States is ~30%, with individuals
born before 1950 having a higher rate of infection than those born later. About
10% of Americans <30 are colonized with the bacteria. This rate of
colonization increases with age, with about 50% of individuals age 50 being
infected. Factors that predispose to higher colonization rates include poor
socioeconomic status and less education. These factors, not race, are
responsible for the rate of H. pylori infection in blacks and Hispanic
Americans being double the rate seen in whites of comparable age. A summary of
risk factors for H. pylori infection is shown in Table 285-2.
Table 285-2: Risk Factors for H. pylori
Infection
|
Transmission
of H. pylori occurs from person to person, following an oral-oral or
fecal-oral route. The risk of H. pylori infection is declining in
developing countries. The rate of infection in the United States has fallen by
>50% when compared to 30 years ago.
Pathophysiology
H. pylori infection is virtually always associated with a
chronic active gastritis, but only 10 to 15% of infected individuals develop
frank peptic ulceration. The basis for this difference is unknown. Initial
studies suggested that >90% of all DUs were associated with H. pylori,
but H. pylori is present in only 30 to 60% of individuals with DU and
70% of patients with GU. The pathophysiology of ulcers not associated with H.
pylori or NSAID ingestion [or the rare Zollinger-Ellison syndrome (ZES)] is
unclear.
The
particular end result of H. pylori infection (gastritis, PUD, gastric
MALT lymphoma, gastric cancer) is determined by a complex interplay between
bacterial and host factors (Fig. 285-6).
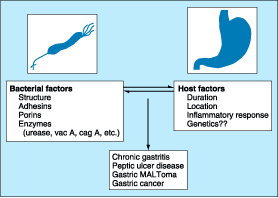
Figure 285-6: Outline of the bacterial and host factors
important in determining H. pylori-induced gastrointestinal disease. MALT,
mucosal-associated lymphoid tissue.
Bacterial factors: H. pylori is able to facilitate gastric
residence, induce mucosal injury, and avoid host defense. Different strains of H.
pylori produce different virulence factors. A specific region of the
bacterial genome, the pathogenicity island, encodes the virulence factors Cag A
and pic B. Vac A also contributes to pathogenicity, though it is not encoded
within the pathogenicity island. These virulence factors, in conjunction with
additional bacterial constituents, can cause mucosal damage. Urease, which
allows the bacteria to reside in the acidic stomach, generates NH3,
which can damage epithelial cells. The bacteria produce surface factors that
are chemotactic for neutrophils and monocytes, which in turn contribute to
epithelial cell injury (see below). H. pylori makes proteases and
phospholipases that break down the glycoprotein lipid complex of the mucous
gel, thus reducing the efficacy of this first line of mucosal defense. H.
pylori expresses adhesins, which facilitate attachment of the bacteria to
gastric epithelial cells. Although lipopolysaccharide (LPS) of gram-negative
bacteria often plays an important role in the infection, H. pylori LPS
has low immunologic activity compared to that of other organisms. It may
promote a smoldering chronic inflammation.
Host factors: The host responds to H. pylori
infection by mounting an inflammatory response, which contributes to gastric
epithelial cell damage without providing immunity against infection. The
neutrophil response is strong both in acute and chronic infection. In addition,
T lymphocytes and plasma cells are components of the chronic inflammatory
infiltrate, supporting the involvement of antigen-specific cellular and humoral
responses. A number of cytokines are released from both epithelial and immune
modulatory cells in response to H. pylori infection including the
proinflammatory cytokines tumor necrosis factor (TNF)![]() ,
interleukin (IL)1
,
interleukin (IL)1![]() /
/![]() ,
IL-6, interferon (IFN)
,
IL-6, interferon (IFN)![]() ,
and granulocyte-macrophage colony stimulating factor. Several chemokines such
as IL-8 and growth-regulated oncogene (GRO)
,
and granulocyte-macrophage colony stimulating factor. Several chemokines such
as IL-8 and growth-regulated oncogene (GRO) ![]() ,
involved in neutrophil recruitment/activation, and RANTES, which recruits
mononuclear cells, have been observed in H. pylori-infected mucosa.
,
involved in neutrophil recruitment/activation, and RANTES, which recruits
mononuclear cells, have been observed in H. pylori-infected mucosa.
The
reason for H. pylori-mediated duodenal ulceration remains unclear. One
potential explanation is that gastric metaplasia in the duodenum of DU patients
permits H. pylori to bind to it and produce local injury secondary to
the host response. Another hypothesis is that H. pylori antral infection
could lead to increased acid production, increased duodenal acid, and mucosal
injury. Basal and stimulated [meal, gastrin-releasing peptide (GRP)] gastrin
release are increased in H. pylori-infected individuals, and
somatostatin-secreting D cells may be decreased. H. pylori infection
might induce increased acid secretion through both direct and indirect actions
of H. pylori and proinflammatory cytokines (IL-8, TNF, and IL-1) on G,
D, and parietal cells (Fig. 285-7). H. pylori infection has also been
associated with decreased duodenal mucosal bicarbonate production. Data
supporting and contradicting each of these interesting theories have been
demonstrated. Thus, the mechanism by which H pylori infection of the
stomach leads to duodenal ulceration remains to be established.
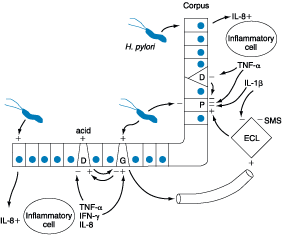
Figure 285-7: Summary of potential mechanisms by which
H. pylori may lead to gastric secretory abnormalities. D, somatostatin cell;
ECL, enterochromaffin-like; G, G cell; IFN, interferon; IL, interleukin; P,
parietal cell; SMS, somatostatin; TNF, tumor necrosis factor.
NSAIDs-induced disease
Epidemiology
NSAIDs
represent one of the most commonly used medications in the United States. More
than 30 billion over-the-counter tablets and 70 million prescriptions are sold
yearly in the United States alone. The spectrum of NSAID-induced morbidity
ranges from nausea and dyspepsia (prevalence reported as high as 50 to 60%) to
a serious gastrointestinal complication such as frank peptic ulceration
complicated by bleeding or perforation in as many as 3 to 4% of users per year.
About 20,000 patients die each year from serious gastrointestinal complications
from NSAIDs. Unfortunately, dyspeptic symptoms do not correlate with
NSAID-induced pathology. Over 80% of patients with serious NSAID-related
complications did not have preceding dyspepsia. In view of the lack of warning
signs, it is important to identify patients who are at increased risk for
morbidity and mortality related to NSAID usage. A summary of established and
possible risk factors is presented in Table 285-3.
Table 285-3: Risk Factors for NSAID-Induced Gastroduodenal Ulceration
|
Pathophysiology
Prostaglandins
play a critical role in maintaining gastroduodenal mucosal integrity and
repair. It therefore follows that interruption of prostaglandin synthesis can
impair mucosal defense and repair, thus facilitating mucosal injury via a
systemic mechanism. A summary of the pathogenetic pathways by which
systemically administered NSAIDs may lead to mucosal injury is shown in Fig.
285-8.
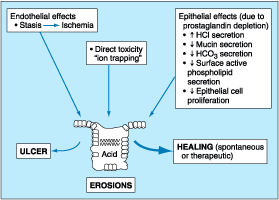
Figure 285-8: Mechanisms by which NSAIDs may induce
mucosal injury.
Injury
to the mucosa also occurs as a result of the topical encounter with NSAIDs.
Aspirin and many NSAIDs are weak acids that remain in a nonionized lipophilic
form when found within the acid environment of the stomach. Under these
conditions, NSAIDs migrate across lipid membranes of epithelial cells, leading
to cell injury once trapped intracellularly in an ionized form. Topical NSAIDs
can also alter the surface mucous layer, permitting back diffusion of H+
and pepsin, leading to further epithelial cell damage.
Miscellaneous Pathogenetic Factors in Acid
Peptic Disease
Cigarette
smoking has been implicated in the pathogenesis of PUD. Not only have smokers
been found to have ulcers more frequently than do nonsmokers, but smoking
appears to decrease healing rates, impair response to therapy, and increase
ulcer-related complications such as perforation. The mechanism responsible for
increased ulcer diathesis in smokers is unknown. Theories have included altered
gastric emptying, decreased proximal duodenal bicarbonate production, and
cigarette-induced generation of noxious mucosal free radicals. Acid secretion
is not abnormal in smokers. Despite these interesting theories, a
unifying mechanism for cigarette-induced peptic ulcer diathesis has not been
established.
Genetic
predisposition has also been considered to play a role in ulcer development.
First-degree relatives of DU patients are three times as likely to develop an
ulcer; however, the potential role of H. pylori infection in contacts is
a major consideration. Increased frequency of blood group O and of the
nonsecretor status have also been implicated as genetic risk factors for peptic
diathesis. However, H. pylori preferentially binds to group O antigens.
Therefore, the role of genetic predisposition in common PUD has not been
established.
Psychological
stress has been thought to contribute to PUD, but studies examining the role of
psychological factors in its pathogenesis have generated conflicting results.
Although PUD is associated with certain personality traits (neuroticism), these
same traits are also present in individuals with nonulcer dyspepsia (NUD) and
other functional and organic disorders. Although more work in this area is
needed, no typical PUD personality has been found.
Diet
has also been thought to play a role in peptic diseases. Certain foods can
cause dyspepsia, but no convincing studies indicate an association between
ulcer formation and a specific diet. This is also true for beverages containing
alcohol and caffeine. Specific chronic disorders have been associated with PUD
(Table 285-4).
Table 285-4: Disorders Associated with Peptic
Ulcer Disease
|
Multiple
factors play a role in the pathogenesis of PUD. The two predominant causes are H.
pylori infection and NSAID ingestion. PUD not related to H. pylori
or NSAIDs may be increasing. Independent of the inciting or injurious agent,
peptic ulcers develop as a result of an imbalance between mucosal
protection/repair and aggressive factors. Gastric acid plays an essential role
in mucosal injury.
Clinical Features
History
Abdominal
pain is common to many gastrointestinal disorders, including DU and GU, but has
a poor predictive value for the presence of either DU or GU. Up to 10% of
patients with NSAID-induced mucosal disease can present with a complication
(bleeding, perforation, and obstruction) without antecedent symptoms. Despite
this poor correlation, a careful history and physical examination are essential
components of the approach to a patient suspected of having peptic ulcers.
Epigastric
pain described as a burning or gnawing discomfort can be present in both DU and
GU. The discomfort is also described as an ill-defined, aching sensation or as
hunger pain. The typical pain pattern in DU occurs 90 min to 3 h after a meal
and is frequently relieved by antacids or food. Pain that awakes the patient
from sleep (between midnight and 3 A.M.) is the most discriminating symptom,
with two-thirds of DU patients describing this complaint. Unfortunately, this
symptom is also present in one-third of patients with NUD. The pain pattern in
GU patients may be different from that in DU patients, where discomfort may
actually be precipitated by food. Nausea and weight loss occur more commonly in
GU patients. In the United States, endoscopy detects ulcers in <30% of
patients who have dyspepsia. Despite this, 40% of these individuals with
typical ulcer symptoms had an ulcer crater, and 40% had gastroduodenitis on
endoscopic examination.
The
mechanism for development of abdominal pain in ulcer patients is unknown.
Several possible explanations include acid-induced activation of chemical
receptors in the duodenum, enhanced duodenal sensitivity to bile acids and
pepsin, or altered gastroduodenal motility.
Variation
in the intensity or distribution of the abdominal pain, as well as the onset of
associated symptoms such as nausea and/or vomiting, may be indicative of an
ulcer complication. Dyspepsia that becomes constant, is no longer relieved by
food or antacids, or radiates to the back may indicate a penetrating ulcer
(pancreas). Sudden onset of severe, generalized abdominal pain may indicate
perforation. Pain worsening with meals, nausea, and vomiting of undigested food
suggest gastric outlet obstruction. Tarry stools or coffee ground emesis
indicate bleeding.
Physical Examination
Epigastric
tenderness is the most frequent finding in patients with GU or DU. Pain may be
found to the right of the midline in 20% of patients. Unfortunately, the
predictive value of this finding is rather low. Physical examination is
critically important for discovering evidence of ulcer complication.
Tachycardia and orthostasis suggest dehydration secondary to vomiting or active
gastrointestinal blood loss. A severely tender, boardlike abdomen suggests a
perforation. Presence of a succussion splash indicates retained fluid in the
stomach, suggesting gastric outlet obstruction.
PUD-Related
Complications
Gastrointestinal Bleeding
Gastrointestinal
bleeding is the most common complication observed in PUD. It occurs in ~15% of
patients and more often in individuals >60 years old. The higher incidence
in the elderly is likely due to the increased use of NSAIDs in this group. As
many as 20% of patients with ulcer-related hemorrhage bleed without any
preceding warning signs or symptoms.
Perforation
The
second most common ulcer-related complication is perforation, being reported in
as many as 6 to 7% of PUD patients. As in the case of bleeding, the incidence
of perforation in the elderly appears to be increasing secondary to increased
use of NSAIDs. Penetration is a form of perforation in which the ulcer bed
tunnels into an adjacent organ. DUs tend to penetrate posteriorly into the
pancreas, leading to pancreatitis, whereas GUs tend to penetrate into the left
hepatic lobe. Gastrocolic fistulas associated with GUs have also been
described.
Gastric Outlet Obstruction
Gastric
outlet obstruction is the least common ulcer-related complication, occurring in
1 to 2% of patients. A patient may have relative obstruction secondary to
ulcer-related inflammation and edema in the peripyloric region. This process
often resolves with ulcer healing. A fixed, mechanical obstruction secondary to
scar formation in the peripyloric areas is also possible. The latter requires
endoscopic (balloon dilation) or surgical intervention. Signs and symptoms
relative to mechanical obstruction may develop insidiously. New onset of early
satiety, nausea, vomiting, increase of postprandial abdominal pain, and weight
loss should make gastric outlet obstruction a possible diagnosis.
Differential Diagnosis
The
list of gastrointestinal and nongastrointestinal disorders that can mimic
ulceration of the stomach or duodenum is quite extensive. The most commonly
encountered diagnosis among patients seen for upper abdominal discomfort is
NUD. NUD, also known as functional dyspepsia or essential dyspepsia,
refers to a group of heterogeneous disorders typified by upper abdominal pain
without the presence of an ulcer. Dyspepsia has been reported to occur in up to
30% of the U.S. population. Up to 60% of patients seeking medical care for
dyspepsia have a negative diagnostic evaluation. The etiology of NUD is not
established, and the potential role of H. pylori in NUD remains
controversial.
Several
additional disease processes that may present with "ulcer-like"
symptoms include proximal gastrointestinal tumors, gastroesophageal reflux,
vascular disease, pancreaticobiliary disease (biliary colic, chronic pancreatitis),
and gastroduodenal Crohn's disease.
Diagnostic Evaluation
In
view of the poor predictive value of abdominal pain for the presence of a
gastroduodenal ulcer and the multiple disease processes that can mimic this
disease, the clinician is often confronted with having to establish the
presence of an ulcer. Documentation of an ulcer requires either a radiographic
(barium study) or an endoscopic procedure.
Barium
studies of the proximal gastrointestinal tract are still commonly used as a
first test for documenting an ulcer. The sensitivity of older single-contrast
barium meals for detecting a DU is as high as 80%, with a double-contrast study
providing detection rates as high as 90%. Sensitivity for detection is
decreased in small ulcers (<0.5 cm), presence of previous scarring, or in
postoperative patients. A DU appears as a well-demarcated crater, most often
seen in the bulb. A GU may represent benign or malignant disease. Typically, a
benign GU also appears as a discrete crater with radiating mucosal folds
originating from the ulcer margin. Ulcers >3 cm in size or those associated
with a mass are more often malignant. Unfortunately, up to 8% of GUs that
appear to be benign by radiographic appearance are malignant by endoscopy or
surgery. Radiographic studies that show a GU must be followed by endoscopy and
biopsy.
Endoscopy
provides the most sensitive and specific approach for examining the upper
gastrointestinal tract. In addition to permitting direct visualization of the
mucosa, endoscopy facilitates photographic documentation of a mucosal defect
and tissue biopsy to rule out malignancy (GU) or H. pylori. Endoscopic
examination is particularly helpful in identifying lesions too small to detect
by radiographic examination, for evaluation of atypical radiographic
abnormalities, or to determine if an ulcer is a source of blood loss.
Although
the methods for diagnosing H. pylori are outlined in Chap. 154, a brief
summary will be included here (Table 285-5). PyloriTek, a biopsy urease test,
has a sensitivity and specificity of >90 to 95%. In the interest of making a
diagnosis of H. pylori without the need for performing endoscopy,
several noninvasive methods for detecting this organism have been developed.
Three types of studies routinely used include serologic testing, the 13C-
or 14C-urea breath test, and the fecal H. pylori antigen
test.
Table 285-5: Tests for Detection of H. pylori
|
||||||||||||||||||||||||||||||
Occasionally,
specialized testing such as serum gastrin and gastric acid analysis or sham
feeding may be needed in individuals with complicated or refractory PUD (see
"Zollinger-Ellison Syndrome," below). Screening for aspirin or NSAIDS
(blood or urine) may also be necessary in refractory, H. pylori-negative
PUD patients.
![]() Treatment
Treatment
Before
the discovery of H. pylori, the therapy of PUD disease was centered on
the old dictum by Schwartz of "no acid, no ulcer." Although acid secretion
is still important in the pathogenesis of PUD, eradication of H. pylori
and therapy/prevention of NSAID-induced disease is the mainstay. A summary of
commonly used drugs for treatment of acid peptic disorders is shown in Table
285-6.
Table 285-6: Drugs Used in the Treatment of
Peptic Ulcer Disease
|
Acid Neutralizing/Inhibitory Drugs
Antacids
Before
we understood the important role of histamine in stimulating parietal cell
activity, neutralization of secreted acid with antacids constituted the main
form of therapy for peptic ulcers. They are now rarely, if ever, used as the
primary therapeutic agent but instead are often used by patients for
symptomatic relief of dyspepsia. The most commonly used agents are mixtures of
aluminum hydroxide and magnesium hydroxide. Aluminum hydroxide can produce
constipation and phosphate depletion; magnesium hydroxide may cause loose stools.
Many of the commonly used antacids (e.g., Maalox, Mylanta) have a combination
of both aluminum and magnesium hydroxide in order to avoid these side effects.
The magnesium-containing preparation should not be used in chronic renal
failure patients because of possible hypermagnesemia, and aluminum may cause
chronic neurotoxicity in these patients.
Calcium
carbonate and sodium bicarbonate are potent antacids with varying levels of
potential problems. The long-term use of calcium carbonate (converts to calcium
chloride in the stomach) can lead to milk-alkali syndrome (hypercalcemia,
hyperphosphatemia with possible renal calcinosis and progression to renal
insufficiency). Sodium bicarbonate may induce systemic alkalosis.
H2 Receptor antagonists
Four
of these agents are presently available (cimetidine, ranitidine, famotidine,
and nizatidine), and their structures share homology with histamine (Fig.
285-9). Although each has different potency, all will significantly inhibit
basal and stimulated acid secretion to comparable levels when used at
therapeutic doses. Moreover, similar ulcer-healing rates are achieved with each
drug when used at the correct dosage. Presently, this class of drug is often
used for treatment of active ulcers (4 to 6 weeks) in combination with
antibiotics directed at eradicating H. pylori (see below).
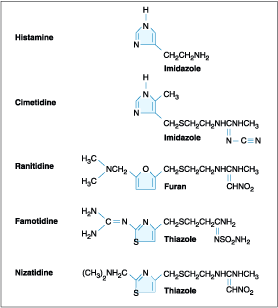
Figure 285-9: Structure of H2 receptor
antagonists.
Cimetidine
was the first H2 receptor antagonist used for the treatment of acid
peptic disorders. The initial recommended dosing profile for cimetidine was 300
mg four times per day. Subsequent studies have documented the efficacy of using
800 mg at bedtime for treatment of active ulcer, with healing rates approaching
80% at 4 weeks. Cimetidine may have weak antiandrogenic side effects resulting
in reversible gynecomastia and impotence, primarily in patients receiving high
doses for prolonged periods of time (months to years, as in ZES). In view of
cimetidine's ability to inhibit cytochrome P450, careful monitoring of drugs
such as warfarin, phenytoin, and theophylline is indicated with long-term
usage. Other rare reversible adverse effects reported with cimetidine include
confusion and elevated levels of serum aminotransferases, creatinine, and serum
prolactin. Ranitidine, famotidine, and nizatidine are more potent H2
receptor antagonists than cimetidine. Each can be used once a day at bedtime.
Comparable nighttime dosing regimens are ranitidine, 300 mg, famotidine, 40 mg,
and nizatidine, 300 mg.
Additional
rare, reversible systemic toxicities reported with H2 receptor
antagonists include pancytopenia, neutropenia, anemia, and thrombocytopenia,
with a prevalence rate varying from 0.01 to 0.2%. Cimetidine and rantidine (to
a lesser extent) can bind to hepatic cytochrome P450, whereas the newer agents,
famotidine and nizatidine, do not.
Proton pump (H+,K+-ATPase)
inhibitors
Omeprazole,
lansoprazole, and the newest additions, rabeprazole and pantoprazole, are
substituted benzimidazole derivatives that covalently bind and irreversibly
inhibit H+,K+-ATPase. These are the most potent acid
inhibitory agents available. Omeprazole and lansoprazole are the proton pump
inhibitors (PPIs) that have been used for the longest time. Both are acid
labile and are administered as enteric-coated granules in a sustained-release
capsule that dissolves within the small intestine at a pH of 6. These agents
are lipophilic compounds; upon entering the parietal cell, they are protonated
and trapped within the acid environment of the tubulovesicular and canalicular
system. These agents potently inhibit all phases of gastric acid secretion.
Onset of action is rapid, with a maximum acid inhibitory effect between 2 and 6
h after administration and duration of inhibition lasting up to 72 to 96 h.
With repeated daily dosing, progressive acid inhibitory effects are observed,
with basal and secretagogue-stimulated acid production being inhibited by
>95% after 1 week of therapy. The half-life of PPIs is approximately 18 h,
thus it can take between 2 and 5 days for gastric acid secretion to return to
normal levels once these drugs have been discontinued. Because the pumps need
to be activated for these agents to be effective, their efficacy is maximized
if they are administered before a meal (e.g., in the morning before breakfast).
Standard dosing for omeprazole and lansoprazole is 20 mg and 30 mg once per
day, respectively. Mild to moderate hypergastrinemia has been observed in
patients taking these drugs. Carcinoid tumors developed in some animals given
the drugs preclinically; however, extensive experience has failed to
demonstrate gastric carcinoid tumor development in humans. Serum gastrin levels
return to normal levels within 1 to 2 weeks after drug cessation. As with any
agent that leads to significant hypochlorhydria, PPIs may interfere with
absorption of drugs such as ketoconazole, ampicillin, iron, and digoxin.
Hepatic cytochrome P450 can be inhibited by these agents, but the overall
clinical significance of this observation is not definitely established.
Caution should be taken when using warfarin, diazepam, and phenytoin
concomitantly with PPIs.
Cytoprotective Agents
Sucralfate
Sucralfate
is a complex sucrose salt in which the hydroxyl groups have been substituted by
aluminum hydroxide and sulfate. This compound is insoluble in water and becomes
a viscous paste within the stomach and duodenum, binding primarily to sites of
active ulceration. Sucralfate may act by several mechanisms. In the gastric
environment, aluminum hydroxide dissociates, leaving the polar sulfate anion,
which can bind to positively charged tissue proteins found within the ulcer
bed, and providing a physicochemical barrier impeding further tissue injury by
acid and pepsin. Sucralfate may also induce a trophic effect by binding growth
factors such as EGF, enhance prostaglandin synthesis, stimulate mucous and
bicarbonate secretion, and enhance mucosal defense and repair. Toxicity from
this drug is rare, with constipation being the most common one reported (2 to
3%). It should be avoided in patients with chronic renal insufficiency to
prevent aluminum-induced neurotoxicity. Hypophosphatemia and gastric bezoar
formation have also been rarely reported. Standard dosing of sucralfate is 1 g
four times per day.
Bismuth-containing preparations
Sir
William Osler considered bismuth-containing compounds the drug of choice for
treating PUD. The resurgence in the use of these agents is due to their effect
against H. pylori. Colloidal bismuth subcitrate (CBS) and bismuth
subsalicylate (BSS, Pepto-Bismol) are the most widely used preparations. The
mechanism by which these agents induce ulcer healing is unclear. Potential
mechanisms include ulcer coating; prevention of further pepsin/HCl-induced
damage; binding of pepsin; and stimulation of prostaglandins, bicarbonate, and
mucous secretion. Adverse effects with short-term usage are rare with bismuth
compounds. Long-term usage with high doses, especially with the avidly absorbed
CBS, may lead to neurotoxicity. These compounds are commonly used as one of the
agents in an anti-H. pylori regimen (see below).
Prostaglandin analogues
In
view of their central role in maintaining mucosal integrity and repair, stable
prostaglandin analogues were developed for the treatment of PUD. The
prostaglandin E1 derivative misoprostal is the only agent of this
class approved by the U.S. Food and Drug Administration for clinical use in the
prevention of NSAID-induced gastroduodenal mucosal injury (see below). The
mechanism by which this rapidly absorbed drug provides its therapeutic effect
is through enhancement of mucosal defense and repair. Prostaglandin analogues
enhance mucous bicarbonate secretion, stimulate mucosal blood flow, and
decrease mucosal cell turnover. The most common toxicity noted with this drug
is diarrhea (10 to 30% incidence). Other major toxicities include uterine
bleeding and contractions; misoprostal is contraindicated in women who may be
pregnant, and women of childbearing age must be made clearly aware of this
potential drug toxicity. The standard therapeutic dose is 200 ![]() g
four times per day.
g
four times per day.
Miscellaneous drugs
A
number of drugs aimed at treating acid peptic disorders have been developed
over the years. In view of their limited utilization in the United States, if
any, they will only be listed briefly. Anticholinergics, designed to inhibit
activation of the muscarinic receptor in parietal cells, met with limited
success due to their relatively weak acid-inhibiting effect and significant
side effects (dry eyes, dry mouth, urinary retention). Tricyclic
antidepressants have been suggested by some, but again the toxicity of these
agents in comparison to the safe, effective drugs already described, precludes
their utility. Finally, the licorice extract carbenoxolone has aldosterone-like
side effects with fluid retention and hypokalemia, making it an undesirable
therapeutic option.
Therapy of H. pylori
Extensive
effort has been placed into determining who of the many individuals with H.
pylori infection should be treated. The common conclusion arrived at by
multiple consensus conferences (National Institutes of Health Consensus
Development, American Digestive Health Foundation International Update
Conference, European Maastricht Consensus, and Asia Pacific Consensus
Conference) is that H. pylori should be eradicated in patients with
documented PUD. This holds true independent of time of presentation (first
episode or not), severity of symptoms, presence of confounding factors such as
ingestion of NSAIDs, or whether the ulcer is in remission. Some have advocated
treating patients with a history of documented PUD who are found to be H.
pylori-positive by serology or breath testing. Over half of patients with
gastric MALT lymphoma experience complete remission of the tumor in response to
H. pylori eradication. Treating patients with NUD or to prevent gastric
cancer remains controversial.
Multiple
drugs have been evaluated in the therapy of H. pylori. No single agent
is effective in eradicating the organism. Combination therapy for 14 days
provides the greatest efficacy. A short-time course administration (7 to 10
days), although attractive, has not proven as successful as the 14-day
regimens. The agents used with the greatest frequency include amoxicillin,
metronidazole, tetracycline, clarithromycin, and bismuth compounds.
The
physician's goal in treating PUD is to provide relief of symptoms (pain or
dyspepsia), promote ulcer healing, and ultimately prevent ulcer recurrence and
complications. The greatest impact of understanding the role of H. pylori
in peptic disease has been the ability to prevent recurrence of what was often
a recurring disease. Documented eradication of H. pylori in patients
with PUD is associated with a dramatic decrease in ulcer recurrence to 4% (as
compared to 59%) in GU patients and 6% (compared to 67%) in DU patients.
Eradication of the organism may lead to diminished recurrent ulcer bleeding.
The impact of its eradication on ulcer perforation is unclear.
Suggested
treatment regimens for H. pylori are outlined in Table 285-7. Choice of
a particular regimen will be influenced by several factors including efficacy,
patient tolerance, existing antibiotic resistance, and cost of the drugs. The
aim for initial eradication rates should be 85 to 90%. Dual therapy [PPI plus
amoxicillin, PPI plus clarithromycin, ranitidine bismuth citrate (Tritec) plus
clarithromycin] are not recommended in view of studies demonstrating
eradication rates of <80 to 85%. The combination of bismuth, metronidazole,
and tetracycline was the first triple regimen found effective against H.
pylori. The combination of two antibiotics plus either a PPI, H2
blocker, or bismuth compound has comparable success rates. Addition of acid
suppression assists in providing early symptom relief and may enhance bacterial
eradication.
Table 285-7: Regimens Recommended for
Eradication of H. pylori Infection
|
|||||||||||||||||||
Triple
therapy, although effective, has several drawbacks, including the potential for
poor patient compliance and drug-induced side effects. Compliance is being
addressed somewhat by simplifying the regimens so that patients can take the
medications twice a day. Simpler (dual therapy) and shorter regimens (7 and 10
days) are not as effective as triple therapy for 14 days. Two anti-H. pylori
regimens are available in prepackaged formulation: Prevpac (lansoprazole,
clarithromycin, and amoxicillin) and Helidac (bismuth subsalicylate,
tetracycline, and metronidazole). The contents of the Prevpac are to be taken
twice per day for 14 days, whereas Helidac constituents are taken four times
per day with an antisecretory agent (PPI or H2 blocker), also taken
for at least 14 days.
Side
effects have been reported in up to 20 to 30% of patients on triple therapy.
Bismuth may cause black stools, constipation, or darkening of the tongue. The
most feared complication with amoxicillin is pseudomembranous colitis, but this
occurs in <1 to 2% of patients. Amoxicillin can also lead to
antibiotic-associated diarrhea, nausea, vomiting, skin rash, and allergic
reaction. Tetracycline has been reported to cause rashes and very rarely
hepatotoxicity and anaphylaxis.
One
important concern with treating patients who may not need treatment is the
potential for development of antibiotic-resistant strains. The incidence and
type of antibiotic-resistant H. pylori strains vary worldwide. Strains
resistant to metronidazole, clarithromycin, amoxicillin, and tetracycline have
been described, with the latter two being uncommon. Antibiotic-resistant
strains are the most common cause for treatment failure in compliant patients.
Unfortunately, in vitro resistance does not predict outcome in patients.
Culture and sensitivity testing of H. pylori is not performed routinely.
Although resistance to metronidazole has been found in as many as 30% and 95%
of isolates in North America and Asia, respectively, triple therapy is
effective in eradicating the organism in >50% of patients infected with a
resistant strain.
Failure
of H. pylori eradication with triple therapy is usually due to infection
with a resistant organism. Quadruple therapy (Table 285-7) where clarithromycin
is substituted for metronidazole (or vice versa) should be the next step. If
eradication is still not achieved in a compliant patient, then culture and sensitivity
of the organism should be considered.
Reinfection
after successful eradication of H. pylori is rare in the United States
(<1%/year). If recurrent infection occurs within the first 6 months after
completing therapy, the most likely explanation is recrudescence as opposed to
reinfection, which occurs later in time.
Therapy of NSAID-Related
Gastric or Duodenal Injury
Medical
intervention for NSAID-related mucosal injury includes treatment of an active
ulcer and prevention of future injury. Recommendations for the treatment and
prevention of NSAID-related mucosal injury are in Table 285-8. Ideally the
injurious agent should be stopped as the first step in the therapy of an active
NSAID-induced ulcer. If that is possible, then treatment with one of the acid
inhibitory agents (H2 blockers, PPIs) is indicated. Cessation of
NSAIDs is not always possible because of the patient's severe underlying
disease. Only PPIs can heal GUs or DUs, independent of whether NSAIDs are
discontinued.
Table 285-8: Recommendations for Treatment of NSAID-Related Mucosal Injury
|
|||||||||||||||
Prevention
of NSAID-induced ulceration can be accomplished by misoprostol (200 ![]() g
qid) or a PPI. High-dose H2 blockers (famotidine, 40 mg bid) have
also shown some promise. The use of COX-2-selective NSAIDs may also reduce
injury to gastric mucosa. Two highly selective COX-2 inhibitors, celecoxib and
rofecoxib, are 100 times more selective inhibitors of COX-2 than standard
NSAIDs, leading to gastric or duodenal mucosal injury that is comparable to
placebo. However, evaluation of possible drug toxicities, such as altered renal
function and induction of thrombosis, requires more data.
g
qid) or a PPI. High-dose H2 blockers (famotidine, 40 mg bid) have
also shown some promise. The use of COX-2-selective NSAIDs may also reduce
injury to gastric mucosa. Two highly selective COX-2 inhibitors, celecoxib and
rofecoxib, are 100 times more selective inhibitors of COX-2 than standard
NSAIDs, leading to gastric or duodenal mucosal injury that is comparable to
placebo. However, evaluation of possible drug toxicities, such as altered renal
function and induction of thrombosis, requires more data.
Approach and Therapy: Summary
Controversy
continues regarding the best approach to the patient who presents with
dyspepsia (Chap. 41). The discovery of H. pylori and its role in
pathogenesis of ulcers has added a new variable to the equation. Previously, if
a patient <50 presented with dyspepsia and without alarming signs or
symptoms suggestive of an ulcer complication or malignancy, an empirical
therapeutic trial with acid suppression was commonly recommended. Although this
approach is practiced by some today, an approach presently gaining approval for
the treatment of patients with dyspepsia is outlined in Fig. 285-10. The
referral to a gastroenterologist is for the potential need of endoscopy and
subsequent evaluation and treatment if the endoscopy is negative.
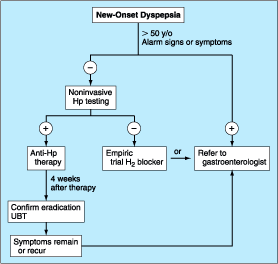
Figure 285-10: Overview of new-onset
dyspepsia. Hp, H. pylori; UBT, urea breath test.
Once
an ulcer (GU or DU) is documented, then the main issue at stake is whether H.
pylori or an NSAID is involved. With H. pylori present, independent
of the NSAID status, triple therapy is recommended for 14 days, followed by
continued acid-suppressing drugs (H2 receptor antagonist or PPIs)
for a total of 4 to 6 weeks. Selection of patients for documentation of H.
pylori eradication is an area of some debate. The test of choice for
documenting eradication is the urea breath test (UBT). The stool antigen study
may also hold promise for this purpose and should certainly be performed if UBT
is not available. Serologic testing is not useful for the purpose of
documenting eradication since antibody titers fall slowly and often do not
become undetectable. Two approaches toward documentation of eradication exist:
(1) test for eradication only in individuals with a complicated course or in
individuals who are frail or with multisystem disease who would do poorly with
an ulcer recurrence, and (2) test all patients for successful eradication. Some
recommend that patients with complicated ulcer disease or who are frail should
be treated with long-term acid suppression, thus making documentation of H.
pylori eradication a moot point. In view of this discrepancy in practice,
it would be best to discuss with the patient the different options available.
Several
issues differentiate the approach to a GU versus a DU. GUs, especially of the
body and fundus, have the potential of being malignant. Multiple biopsies of a
GU should be taken initially; even if these are negative for neoplasm, repeat
endoscopy to document healing at 8 to 12 weeks should be performed, with biopsy
if the ulcer is still present. About 70% of GUs eventually found to be
malignant undergo significant (usually incomplete) healing.
The
majority (>90%) of GUs and DUs heal with the conventional therapy outlined
above. A GU that fails to heal after 12 weeks and a DU that doesn't heal after
8 weeks of therapy should be considered refractory. Once poor compliance and
persistent H. pylori infection have been excluded, NSAID use, either
inadvertent or surreptitious, must be excluded. In addition, cigarette smoking
must be eliminated. For a GU, malignancy must be meticulously excluded. Next,
consideration should be given to a gastric hypersecretory state, which can be
excluded with gastric acid analysis. Although a subset of patients have gastric
acid hypersecretion of unclear etiology as a contributing factor to refractory
ulcers, ZES should be excluded with a fasting gastrin or secretin stimulation
test (see below). More than 90% of refractory ulcers (either DUs or GUs) heal
after 8 weeks of treatment with higher doses of PPI (omeprazole, 40 mg/d). This
higher dose is also effective in maintaining remission. Surgical intervention
may be a consideration at this point; however, other rare causes of refractory
ulcers must be excluded before recommending surgery. Rare etiologies of
refractory ulcers that may be diagnosed by gastric or duodenal biopsies
include: ischemia, Crohn's disease, amyloidosis, sarcoidosis, lymphoma,
eosinophilic gastroenteritis, or infection [cytomegalovirus (CMV),
tuberculosis, or syphilis].
Surgical Therapy
Surgical
intervention in PUD can be viewed as being either elective, for treatment of
medically refractory disease, or as urgent/emergent, for the treatment of an
ulcer-related complication. Refractory ulcers are an exceedingly rare
occurrence. Surgery is more often required for treatment of an ulcer-related
complication. Gastrointestinal bleeding (Chap. 44), perforation, and gastric
outlet obstruction are the three complications that may require surgical
intervention.
Hemorrhage
is the most common ulcer-related complication, occurring in ~15 to 25% of
patients. Bleeding may occur in any age group but is most often seen in older
patients (sixth decade or beyond). The majority of patients stop bleeding
spontaneously, but in some, endoscopic therapy (Chap. 283) is necessary.
Patients unresponsive or refractory to endoscopic intervention will require
surgery (~5% of transfusion-requiring patients).
Free
peritoneal perforation occurs in ~2 to 3% of DU patients. As in the case of
bleeding, up to 10% of these patients will not have antecedent ulcer symptoms.
Concomitant bleeding may occur in up to 10% of patients with perforation, with
mortality being increased substantially. Peptic ulcer can also penetrate into
adjacent organs, especially with a posterior DU, which can penetrate into the
pancreas, colon, liver, or biliary tree.
Pyloric
channel ulcers or DUs can lead to gastric outlet obstruction in ~2 to 3% of
patients. This can result from chronic scarring or from impaired motility due
to inflammation and/or edema with pylorospasm. Patients may present with early
satiety, nausea, vomiting of undigested food, and weight loss. Conservative
management with nasogastric suction, intravenous hydration/nutrition, and
antisecretory agents is indicated for 7 to 10 days with the hope that a
functional obstruction will reverse. If a mechanical obstruction persists,
endoscopic intervention with balloon dilation may be effective. Surgery should
be considered if all else fails.
Specific Operations for Duodenal Ulcers
Surgical
treatment is designed to decrease gastric acid secretion. Operations most
commonly performed include vagotomy and drainage (by pyloroplasty,
gastroduodenostomy, or gastrojejunostomy), highly selective vagotomy (which
does not require a drainage procedure), and vagotomy with antrectomy. The
specific procedure performed is dictated by the underlying circumstances:
elective vs. emergency, the degree and extent of duodenal ulceration, and the
expertise of the surgeon.
Vagotomy
is a component of each of these procedures and is aimed at decreasing acid
secretion through ablating cholinergic input to the stomach. Unfortunately,
both truncal and selective vagotomy (preserves the celiac and hepatic branches)
result in gastric atony despite successful reduction of both basal acid output
(BAO, decreased by 85%) and maximal acid output (MAO, decreased by 50%).
Drainage procedure through pyloroplasty or gastroduodenostomy is required in an
effort to compensate for the vagotomy-induced gastric motility disorder. To
minimize gastric dysmotility, highly selective vagotomy (also known as parietal
cell, super selective, and proximal vagotomy) was developed. Only the vagal
fibers innervating the portion of the stomach that contains parietal cells is
transected, thus leaving fibers important for regulating gastric motility
intact. Although this procedure leads to an immediate decrease in both BAO and
stimulated acid output, acid secretion recovers over time. By the end of the
first postoperative year, basal and stimulated acid output are ~30 and 50%,
respectively, of preoperative levels. Ulcer recurrence rates are higher with
highly selective vagotomy, although the overall complication rates are lower
(Table 285-9).
Table 285-9: Outcome in Patients After
Acid-Reducing Gastric Surgery
|
The
procedure that provides the lowest rates of ulcer recurrence but has the
highest complication rate is vagotomy (truncal or selective) in combination
with antrectomy. Antrectomy is aimed at eliminating an additional stimulant of
gastric acid secretion, gastrin. Gastrin originates from G cells found in the
antrum. Two principal types of reanastomoses are used after antrectomy, gastroduodenostomy
(Billroth I) or gastrojejunostomy (Billroth II) (Fig. 285-11). Although
Billroth I is often preferred over II, severe duodenal inflammation or scarring
may preclude its performance.
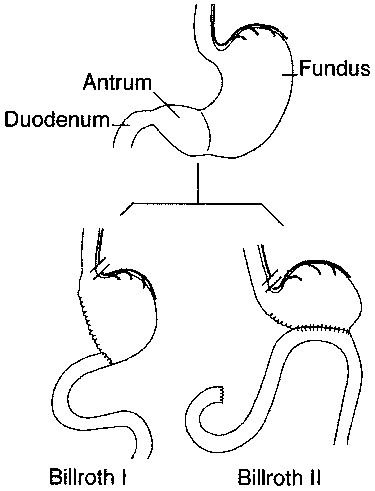
Figure 285-11: Schematic representation
of Billroth I and II procedures.
Of
these procedures, highly selective vagotomy may be the one of choice in the
elective setting, except in situations where ulcer recurrence rates are high
(prepyloric ulcers and those refractory to H2 therapy). Selection of
vagotomy and antrectomy may be more appropriate in these circumstances.
These
procedures have been traditionally performed by standard laparotomy. The advent
of laparoscopic surgery has led several surgical teams to successfully perform
highly selective vagotomy, truncal vagotomy/pyloroplasty, and truncal
vagotomy/antrectomy through this approach. An increase in the number of
laparoscopic procedures for treatment of PUD is expected.
Specific Operations for Gastric Ulcers
The
location and the presence of a concomitant DU dictate the operative procedure
performed for a GU. Antrectomy (including the ulcer) with a Billroth I
anastomosis is the treatment of choice for an antral ulcer. Vagotomy is
performed only if a DU is present. Although ulcer excision with vagotomy and
drainage procedure has been proposed, the higher incidence of ulcer recurrence
makes this a less desirable approach. Ulcers located near the esophagogastric
junction may require a more radical approach, a subtotal gastrectomy with a
Roux-en-Y esophagogastrojejunostomy (Csende's procedure). A less aggressive
approach including antrectomy, intraoperative ulcer biopsy, and vagotomy
(Kelling-Madlener procedure) may be indicated in fragile patients with a high
GU. Ulcer recurrence approaches 30% with this procedure.
Surgery-Related Complications
Complications
seen after surgery for PUD are related primarily to the extent of the
anatomical modification performed. Minimal alteration (highly selective
vagotomy) is associated with higher rates of ulcer recurrence and less
gastrointestinal disturbance. More aggressive surgical procedures have a lower
rate of ulcer recurrence but a greater incidence of gastrointestinal
dysfunction. Overall, morbidity and mortality related to these procedures are
quite low. Morbidity associated with vagotomy and antrectomy or pyloroplasty is
![]() 5%,
with mortality ~1%. Highly selective vagotomy has lower morbidity and mortality
rates of 1 and 0.3%, respectively.
5%,
with mortality ~1%. Highly selective vagotomy has lower morbidity and mortality
rates of 1 and 0.3%, respectively.
In
addition to the potential early consequences of any intraabdominal procedure
(bleeding, infection, thromboembolism), gastroparesis, duodenal stump leak, and
efferent loop obstruction can be observed.
Recurrent Ulceration
The
risk of ulcer recurrence is directly related to the procedure performed (Table
285-9). Ulcers that recur after partial gastric resection tend to develop at
the anastomosis (stomal or marginal ulcer). Epigastric abdominal pain is the
most frequent presenting complaint. Severity and duration of pain tend to be
more progressive than observed with DUs before surgery.
Ulcers
may recur for several reasons including incomplete vagotomy, retained antrum,
and, less likely, persistent or recurrent H. pylori infection. ZES
should have been excluded preoperatively. More recently, surreptitious use of
NSAIDs has been found to be a reason for recurrent ulcers after surgery,
especially if the initial procedure was done for an NSAID-induced ulcer. Once H.
pylori and NSAIDs have been excluded as etiologic factors, the question of
incomplete vagotomy or retained gastric antrum should be explored. For the
latter, fasting plasma gastrin levels should be determined. If elevated,
retained antrum or ZES (see below) should be considered. A combination of acid
secretory analysis and secretin stimulation (see below) can assist in this differential
diagnosis. Incomplete vagotomy can be ruled out by gastric acid analysis
coupled with sham feeding. In this test, gastric acid output is measured while
the patient sees, smells, and chews a meal (without swallowing). The cephalic
phase of gastric secretion, which is mediated by the vagus, is being assessed
with this study. An increase in gastric acid output in response to sham feeding
is evidence that the vagus nerve is intact.
Medical
therapy with H2 blockers will heal postoperative ulceration in 70 to
90% of patients. The efficacy of PPIs has not been fully assessed in this
group, but one may anticipate greater rates of ulcer healing compared to those
obtained with H2 blockers. Repeat operation (complete vagotomy,
partial gastrectomy) may be required in a small subgroup of patients who have
not responded to aggressive medical management.
Afferent Loop Syndromes
Two
types of afferent loop syndrome can occur in patients who have undergone
partial gastric resection with Billroth II anastomosis. The most common of the
two is bacterial overgrowth in the afferent limb secondary to stasis. Patients
may experience postprandial abdominal pain, bloating, and diarrhea with
concomitant malabsorption of fats and vitamin B12. Cases refractory
to antibiotics may require surgical revision of the loop. The less common
afferent loop syndrome can present with severe abdominal pain and bloating that
occur 20 to 60 min after meals. Pain is often followed by nausea and vomiting
of bile-containing material. The pain and bloating may improve after emesis.
The cause of this clinical picture is theorized to be incomplete drainage of
bile and pancreatic secretions from an afferent loop that is partially
obstructed. Cases refractory to dietary measures may need surgical revision.
Dumping Syndrome
Dumping
syndrome consists of a series of vasomotor and gastrointestinal signs and
symptoms and occurs in patients who have undergone vagotomy and drainage
(especially Billroth procedures). Two phases of dumping, early and late, can
occur. Early dumping takes place 15 to 30 min after meals and consists of
crampy abdominal discomfort, nausea, diarrhea, belching, tachycardia,
palpitations, diaphoresis, light-headedness, and, rarely, syncope. These signs
and symptoms arise from the rapid emptying of hyperosmolar gastric contents
into the small intestine, resulting in a fluid shift into the gut lumen with
plasma volume contraction and acute intestinal distention. Release of
vasoactive gastrointestinal hormones (vasoactive intestinal polypeptide,
neurotensin, motilin) is also theorized to play a role in early dumping.
The
late phase of dumping typically occurs 90 min to 3 h after meals. Vasomotor
symptoms (light-headedness, diaphoresis, palpitations, tachycardia, and
syncope) predominate during this phase. This component of dumping is thought to
be secondary to hypoglycemia from excessive insulin release.
Dumping
syndrome is most noticeable after meals rich in simple carbohydrates
(especially sucrose) and high osmolarity. Ingestion of large amounts of fluids
may also contribute. Up to 50% of postvagotomy and drainage patients will
experience dumping syndrome to some degree. Signs and symptoms often improve
with time, but a severe protracted picture can occur in up to 1% of patients.
Dietary
modification is the cornerstone of therapy for patients with dumping syndrome.
Small, multiple (six) meals devoid of simple carbohydrates coupled with
elimination of liquids during meals is important. Antidiarrheals and
anticholinergic agents are complimentary to diet. The somatostatin analogue
octreotide has been successful in diet refractory cases. This drug is
administered subcutaneously (50 ![]() g
tid), titrated according to clinical response. Recently a long-acting
formulation has become available, but its use in dumping syndrome has not been
examined.
g
tid), titrated according to clinical response. Recently a long-acting
formulation has become available, but its use in dumping syndrome has not been
examined.
Postvagotomy Diarrhea
Up
to 10% of patients may seek medical attention for the treatment of postvagotomy
diarrhea. This complication is most commonly observed after truncal vagotomy.
Patients may complain of intermittent diarrhea that occurs typically 1 to 2 h
after meals. Occasionally the symptoms may be severe and relentless. This is
due to a motility disorder from interruption of the vagal fibers supplying the
luminal gut. Other contributing factors may include decreased absorption of
nutrients (see below), increased excretion of bile acids, and release of
luminal factors that promote secretion. Diphenoxylate or loperamide is often
useful in symptom control. The bile salt-binding agent cholestyramine may be
helpful in severe cases. Surgical reversal of a 10-cm segment of jejunum may
yield a substantial improvement in bowel frequency in a subset of patients.
Bile Reflux Gastropathy
A
subset of post-partial gastrectomy patients will present with abdominal pain,
early satiety, nausea, and vomiting, who have as the only finding mucosal
erythema of the gastric remnant. Histologic examination of the gastric mucosa
reveals minimal inflammation but the presence of epithelial cell injury. This
clinical picture is categorized as bile or alkaline reflux
gastropathy/gastritis. Although reflux of bile is implicated as the reason for
this disorder, the mechanism is unknown. Prokinetic agents (cisapride, 10 to 20
mg before meals and at bedtime) and cholestyramine have been effective
treatments. Cisapride may cause cardiac arrhythmias. Severe refractory symptoms
may require using either nuclear scanning with 99mTc-HIDA, to
document reflux, or an alkaline challenge test, where 0.1 N NaOH is
infused into the stomach in an effort to reproduce the patient's symptoms.
Surgical diversion of pancreaticobiliary secretions away from the gastric
remnant with a Roux-en-Y gastrojejunostomy consisting of a long (50 to 60 cm)
Roux limb has been used in severe cases. Bilious vomiting improves, but early
satiety and bloating may persist in up to 50% of patients.
Maldigestion and Malabsorption
Weight
loss can be observed in up to 60% of patients after partial gastric resection.
A significant component of this weight reduction is due to decreased oral
intake. However, mild steatorrhea can also develop. Reasons for
maldigestion/malabsorption include decreased gastric acid production, rapid
gastric emptying, decreased food dispersion in the stomach, reduced luminal
bile concentration, reduced pancreatic secretory response to feeding, and rapid
intestinal transit.
Decreased
serum vitamin B12 levels can be observed after partial gastrectomy.
This is usually not due to deficiency of intrinsic factor (IF), since a minimal
amount of parietal cells (source of IF) are removed during antrectomy. Reduced
vitamin B12 may be due to competition for the vitamin by bacterial
overgrowth or inability to split the vitamin from its protein-bound source due
to hypochlorhydria.
Iron-deficiency
anemia may be a consequence of impaired absorption of dietary iron in patients
with a Billroth II gastrojejunotomy. Absorption of iron salts is normal in
these individuals; thus a favorable response to oral iron supplementation can
be anticipated. Folate deficiency with concomitant anemia can also develop in
these patients. This deficiency may be secondary to decreased absorption or
diminished oral intake.
Malabsorption
of vitamin D and calcium resulting in osteoporosis and osteomalacia is common
after partial gastrectomy and gastrojejunostomy (Billroth II). Osteomalacia can
occur as a late complication in up to 25% of post-partial gastrectomy patients.
Bone fractures occur twice as commonly in men after gastric surgery as in a
control population. It may take years before x-ray findings demonstrate
diminished bone density. Elevated alkaline phosphatase, reduced serum calcium,
bone pain, and pathologic fractures may be seen in patients with osteomalacia.
The high incidence of these abnormalities in this subgroup of patients
justifies treating them with vitamin D and calcium supplementation
indefinitely. Therapy is especially important in females.
Gastric Adenocarcinoma
The
incidence of adenocarcinoma in the gastric stump is increased 15 years after
resection. Some have reported a four- to fivefold increase in gastric cancer 20
to 25 years after resection. The pathogenesis is unclear but may involve
alkaline reflux, bacterial proliferation, or hypochlorhydria. Endoscopic
screening every other year may detect surgically treatable disease.
Related Conditions
Zollinger-Ellison Syndrome
Severe
peptic ulcer diathesis secondary to gastric acid hypersecretion due to unregulated
gastrin release from a non-![]() cell endocrine tumor (gastrinoma) defines the components of the ZES. Initially,
ZES was typified by aggressive and refractory ulceration in which total
gastrectomy provided the only chance for enhancing survival. Today ZES can be
cured by surgical resection in up to 30% of patients.
cell endocrine tumor (gastrinoma) defines the components of the ZES. Initially,
ZES was typified by aggressive and refractory ulceration in which total
gastrectomy provided the only chance for enhancing survival. Today ZES can be
cured by surgical resection in up to 30% of patients.
Epidemiology
The
incidence of ZES varies from 0.1 to 1% of individuals presenting with PUD.
Males are more commonly affected than females, and the majority of patients are
diagnosed between ages 30 and 50. Gastrinomas are classified into sporadic
tumors (more common) and those associated with multiple endocrine neoplasia
(MEN) type I (see below).
Pathophysiology
Hypergastremia
originating from an autonomous neoplasm is the driving force responsible for
the clinical manifestations in ZES. Gastrin stimulates acid secretion through
gastrin receptors on parietal cells and by inducing histamine release from ECL
cells. Gastrin also has a trophic action on gastric epithelial cells.
Longstanding hypergastrinemia leads to markedly increased gastric acid
secretion through both parietal cell stimulation and increased parietal cell
mass. The increased gastric acid output leads to the peptic ulcer diathesis,
erosive esophagitis, and diarrhea.
Tumor Distribution
Although
early studies suggested that the vast majority of gastrinomas occurred within
the pancreas, a significant number of these lesions are extrapancreatic. Over
80% of these tumors are found within the hypothetical gastrinoma triangle
(confluence of the cystic and common bile ducts superiorly, junction of the
second and third portions of the duodenum inferiorly, and junction of the neck
and body of the pancreas medially). Duodenal tumors constitute the most common
nonpancreatic lesion; up to 50% of gastrinomas are found here. Less common
extrapancreatic sites include stomach, bones, ovaries, heart, liver, and lymph
nodes. More than 60% of tumors are considered malignant, with up to 30 to 50%
of patients having multiple lesions or metastatic disease at presentation.
Histologically, gastrin-producing cells appear well differentiated, expressing
markers typically found in endocrine neoplasms (chromogranin, neuron-specific
enolase).
Clinical Manifestations
Gastric
acid hypersecretion is responsible for the signs and symptoms observed in
patients with ZES. Peptic ulcer is the most common clinical manifestation,
occurring in >90% of gastrinoma patients. Initial presentation and ulcer
location (duodenal bulb) may be indistinguishable from common PUD. Clinical
situations that should create suspicion of gastrinoma are ulcers in unusual
locations (second part of the duodenum and beyond), ulcers refractory to
standard medical therapy, ulcer recurrence after acid-reducing surgery, or
ulcers presenting with frank complications (bleeding, obstruction, and
perforation). Symptoms of esophageal origin are present in up to two-thirds of
patients with ZES, with a spectrum ranging from mild esophagitis to frank
ulceration with stricture and Barrett's mucosa.
Diarrhea
is the next most common clinical manifestation in up to 50% of patients.
Although diarrhea often occurs concomitantly with acid peptic disease, it may
also occur independent of an ulcer. Etiology of the diarrhea is multifactorial,
resulting from marked volume overload to the small bowel, pancreatic enzyme
inactivation by acid, and damage of the intestinal epithelial surface by acid.
The epithelial damage can lead to a mild degree of maldigestion and
malabsorption of nutrients. The diarrhea may also have a secretory component
due to the direct stimulatory effect of gastrin on enterocytes or the
cosecretion of additional hormones from the tumor, such as vasoactive
intestinal peptide.
Gastrinomas
can develop in the presence of MEN I syndrome (Chap. 93) in approximately 25%
of patients. This autosomal dominant disorder involves primarily three organ
sites: the parathyroid glands (80 to 90%), pancreas (40 to 80%), and pituitary
gland (30 to 60%). The genetic defect in MEN I is in the long arm of chromosome
11 (11q11-q13). In view of the stimulatory effect of calcium on gastric
secretion, the hyperparathyroidism and hypercalcemia seen in MEN I patients may
have a direct effect on ulcer disease. Resolution of hypercalcemia by
parathyroidectomy reduces gastrin and gastric acid output in gastrinoma
patients. An additional distinguishing feature in ZES patients with MEN I is
the higher incidence of gastric carcinoid tumor development (as compared to
patients with sporadic gastrinomas). Gastrinomas tend to be smaller, multiple,
and located in the duodenal wall more often than is seen in patients with
sporadic ZES. Establishing the diagnosis of MEN I is critical not only from the
standpoint of providing genetic counseling to the patient and his or her family
but also from the surgical approach recommended.
Diagnosis
The
first step in the evaluation of a patient suspected of having ZES is to obtain
a fasting gastrin level. A list of clinical scenarios that should arouse
suspicion regarding this diagnosis is shown in Table 285-10. Fasting gastrin
levels are usually <150 pg/mL. Virtually all gastrinoma patients will have a
gastrin level >150 to 200 pg/mL. Measurement of fasting gastrin should be
repeated to confirm the clinical suspicion.
Table 285-10: When to Obtain a Fasting Serum
Gastrin Level
|
Multiple
processes can lead to an elevated fasting gastrin level (Table 285-11), with
gastric hypochlorhydria or achlorhydria being the most frequent causes. Gastric
acid induces feedback inhibition of gastrin release. A decrease in acid
production will subsequently lead to failure of the feedback inhibitory
pathway, resulting in net hypergastrinemia. Gastrin levels will thus be high in
patients using antisecretory agents for the treatment of acid peptic disorders
and dyspepsia. H. pylori infection can also cause hypergastrinemia.
Table 285-11: Differential Diagnosis of
Hypergastrinemia
|
The
next step in establishing a biochemical diagnosis of gastrinoma is to assess
acid secretion. Nothing further needs to be done if decreased acid output is
observed. In contrast, normal or elevated gastric acid output suggests a need
for additional tests. Gastric acid analysis is performed by placing a
nasogastric tube in the stomach and drawing samples at 15-min intervals for 1 h
during unstimulated or basal state (BAO), followed by continued sampling after
administration of intravenous pentagastrin (MAO). Up to 90% of gastrinoma
patients may have a BAO of ![]() 15
meq/h (normal <4 meq/h). Up to 12% of patients with common PUD may have
comparable levels of acid secretion. A BAO/MAO ratio >0.6 is highly
suggestive of ZES, but a ratio <0.6 does not exclude the diagnosis.
15
meq/h (normal <4 meq/h). Up to 12% of patients with common PUD may have
comparable levels of acid secretion. A BAO/MAO ratio >0.6 is highly
suggestive of ZES, but a ratio <0.6 does not exclude the diagnosis.
Gastrin
provocative tests have been developed in an effort to differentiate between the
causes of hypergastrinemia and are especially helpful in patients with
indeterminant acid secretory studies. The tests are the secretin stimulation
test, the calcium infusion study, and a standard meal test. In each of these, a
fasted patient has an indwelling intravenous catheter in place for serial blood
sampling and an intravenous line in place for secretin or calcium infusion. The
patient receives either secretion (intravenous bolus of 2 ![]() g/kg)
or calcium (calcium gluconate, 5 mg/kg body weight over 3 h) or is fed a meal.
Blood is then drawn at predetermined intervals (10 min and 1 min before and at
2, 5, 10, 15, 20, and 30 min after injection for secretin stimulation and at
30-min intervals during the calcium infusion). The most sensitive and specific
gastrin provocative test for the diagnosis of gastrinoma is the secretin study.
An increase in gastrin of
g/kg)
or calcium (calcium gluconate, 5 mg/kg body weight over 3 h) or is fed a meal.
Blood is then drawn at predetermined intervals (10 min and 1 min before and at
2, 5, 10, 15, 20, and 30 min after injection for secretin stimulation and at
30-min intervals during the calcium infusion). The most sensitive and specific
gastrin provocative test for the diagnosis of gastrinoma is the secretin study.
An increase in gastrin of ![]() 200
pg within 15 min of secretin injection has a sensitivity and specificity of
>90% for ZES. The calcium infusion study is less sensitive and specific than
the secretin test, with a rise of >400 pg/mL observed in ~80% of gastrinoma
patients. The lower accuracy, coupled with it being a more cumbersome study
with greater potential for adverse effects, makes calcium infusion less useful
and therefore rarely, if ever, utilized. Rarely, one may observe increased BAO
and hypergastrinemia in a patient who in the past has been categorized as
having G cell hyperplasia or hyperfunction. This set of findings may have been
due to H. pylori. The standard meal test was devised to assist in making
the diagnosis of G cell-related hyperactivity, by observing a dramatic increase
in gastrin after a meal (>200%). This test is not useful in differentiating
between G cell hyperfunction and ZES.
200
pg within 15 min of secretin injection has a sensitivity and specificity of
>90% for ZES. The calcium infusion study is less sensitive and specific than
the secretin test, with a rise of >400 pg/mL observed in ~80% of gastrinoma
patients. The lower accuracy, coupled with it being a more cumbersome study
with greater potential for adverse effects, makes calcium infusion less useful
and therefore rarely, if ever, utilized. Rarely, one may observe increased BAO
and hypergastrinemia in a patient who in the past has been categorized as
having G cell hyperplasia or hyperfunction. This set of findings may have been
due to H. pylori. The standard meal test was devised to assist in making
the diagnosis of G cell-related hyperactivity, by observing a dramatic increase
in gastrin after a meal (>200%). This test is not useful in differentiating
between G cell hyperfunction and ZES.
Tumor Localization
Once
the biochemical diagnosis of gastrinoma has been confirmed, the tumor must be
located. Multiple imaging studies have been utilized in an effort to enhance
tumor localization (Table 285-12). The broad range of sensitivity is due to the
variable success rates achieved by the different investigative groups.
Endoscopic ultrasound (EUS) permits imaging of the pancreas with a high degree
of resolution (<5 mm). This modality is particularly helpful in excluding
small neoplasms within the pancreas and in assessing the presence of
surrounding lymph nodes and vascular involvement. Several types of endocrine
tumors express cell-surface receptors for somatostatin. This permits the
localization of gastrinomas by measuring the uptake of the stable somatostatin
analogue, 111In-pentriotide (octreoscan) with sensitivity and
specificity rates of >75%.
Table 285-12: Sensitivity of Imaging Studies in
Zollinger-Ellison Syndrome
|
||||||||||||||||||||||||||||||||||||
Up
to 50% of patients have metastatic disease at diagnosis. Success in controlling
gastric acid hypersecretion has shifted the emphasis of therapy towards
providing a surgical cure. Detecting the primary tumor and excluding metastatic
disease are critical in view of this paradigm shift. Once a biochemical
diagnosis has been confirmed, the patient should first undergo an abdominal
computed tomographic scan, magnetic resonance imaging, or octreoscan (depending
on availability) to exclude metastatic disease. Once metastatic disease has
been excluded, an experienced endocrine surgeon may opt for exploratory
laparotomy with intraoperative ultrasound or transillumination. In other
centers, careful examination of the peripancreatic area with EUS, accompanied
by endoscopic exploration of the duodenum for primary tumors, will be performed
before surgery. Selective arterial secretin injection (SASI) may be a useful
adjuvant for localizing tumors in a subset of patients.
![]() Treatment
Treatment
Treatment
of functional endocrine tumors is directed at ameliorating the signs and
symptoms related to hormone overproduction, curative resection of the neoplasm,
and attempts to control tumor growth in metastatic disease.
PPIs
are the treatment of choice and have decreased the need for total gastrectomy.
Initial doses of omeprazole or lansoprazole should be in the range of 60 mg/d.
Dosing can be adjusted to achieve a BAO <10 meq/h (at the drug trough) in
surgery-naive patients and to <5 meq/h in individuals who have previously
undergone an acid-reducing operation. Although the somatostatin analogue has
inhibitory effects on gastrin release from receptor-bearing tumors and inhibits
gastric acid secretion to some extent, PPIs have the advantage of reducing
parietal cell activity to a greater degree.
The
ultimate goal of surgery would be to provide a definitive cure. Improved
understanding of tumor distribution has led to 10-year disease-free intervals
as high as 34% in sporadic gastrinoma patients undergoing surgery. A positive
outcome is highly dependent on the experience of the surgical team treating
these rare tumors. Surgical therapy of gastrinoma patients with MEN I remains
controversial because of the difficulty in rendering these patients disease
free with surgery. In contrast to the encouraging postoperative results
observed in patients with sporadic disease, only 6% of MEN I patients are
disease free 5 years after an operation. Some groups suggest surgery only if a
clearly identifiable, nonmetastatic lesion is documented by structural studies.
Others advocate a more aggressive approach, where all patients free of hepatic
metastasis are explored and all detected tumors in the duodenum are resected;
this is followed by enucleation of lesions in the pancreatic head, with a
distal pancreatectomy to follow. The outcome of the two approaches has not been
clearly defined.
Therapy
of metastatic endocrine tumors in general remains suboptimal; gastrinomas are
no exception. A host of medical therapeutic approaches including chemotherapy
(streptozotocin, 5-fluorouracil, and doxorubicin), IFN-![]() ,
and hepatic artery embolization lead to significant toxicity without a substantial
improvement in overall survival. Surgical approaches including debulking
surgery and liver transplantation for hepatic metastasis have also produced
limited benefit. Therefore, early recognition and surgery are the only chances
for curing this disease.
,
and hepatic artery embolization lead to significant toxicity without a substantial
improvement in overall survival. Surgical approaches including debulking
surgery and liver transplantation for hepatic metastasis have also produced
limited benefit. Therefore, early recognition and surgery are the only chances
for curing this disease.
The
5- and 10-year survival rates for gastrinoma patients are 62 to 75% and 47 to
53%, respectively. Individuals with the entire tumor resected or those with a
negative laparotomy have 5- and 10-year survival rates >90%. Patients with
incompletely resected tumors have 5- and 10-year survival of 43% and 25%,
respectively. Patients with hepatic metastasis have <20% survival at 5
years. Favorable prognostic indicators include primary duodenal wall tumors,
isolated lymph node tumor, and undetectable tumor upon surgical exploration.
Poor prognostic indicators include hepatic metastases or the presence of
Cushing's syndrome in a sporadic gastrinoma patient.
Stress-Related Mucosal Injury
Patients
suffering from shock, sepsis, massive burns, severe trauma, or head injury can
develop acute erosive gastric mucosal changes or frank ulceration with
bleeding. Classified as stress-induced gastritis or ulcers, injury is most
commonly observed in the acid-producing (fundus and body) portions of the
stomach. The most common presentation is gastrointestinal bleeding, which is
usually minimal but can occasionally be life-threatening. Respiratory failure
requiring mechanical ventilation and underlying coagulopathy are risk factors
for bleeding, which tends to occur 48 to 72 h after the acute injury or insult.
Histologically,
stress injury does not contain inflammation or H. pylori; thus
"gastritis" is a misnomer. Although elevated gastric acid secretion
may be noted in patients with stress ulceration after head trauma (Cushing's
ulcer) and severe burns (Curling's ulcer), mucosal ischemia and breakdown of
the normal protective barriers of the stomach also play an important role in
the pathogenesis. Acid must contribute to injury in view of the significant
drop in bleeding noted when acid inhibitors are used as a prophylactic measure
for stress gastritis.
Improvement
in the general management of intensive care unit patients has led to a
significant decrease in the incidence of gastrointestinal bleeding due to
stress ulceration. The estimated decrease in bleeding is from 20 to 30% to
<15%. This improvement has led to some debate regarding the need for
prophylactic therapy. The limited benefit of medical (endoscopic, angiographic)
and surgical therapy in a patient with hemodynamically compromising bleeding
associated with stress ulcer/gastritis supports the use of preventive measures
in high-risk patients (mechanically ventilated, coagulopathy, multiorgan
failure, or severe burns). Maintenance of gastric pH >3.5 with continuous infusion
of H2 blockers or liquid antacids administered every 2 to 3 h are
viable options. Sucralfate slurry (1 g every 4 to 6 h) has also been
successful. If bleeding occurs despite these measures, endoscopy, intraarterial
vasopressin, or embolization are options. If all else fails, then surgery
should be considered. Although vagotomy and antrectomy may be used, the better
approach would be a total gastrectomy, which has an exceedingly high mortality
rate in this setting.
Gastritis
The
term gastritis should be reserved for histologically documented
inflammation of the gastric mucosa. Gastritis is not the mucosal
erythema seen during endoscopy and is not interchangeable with
"dyspepsia." The etiologic factors leading to gastritis are broad and
heterogeneous. Gastritis has been classified based on time course (acute vs.
chronic), histologic features, and anatomic distribution or proposed pathogenic
mechanism (Table 285-13).
Table 285-13: Classification of Gastritis
|
The
correlation between the histologic findings of gastritis, the clinical picture
of abdominal pain or dyspepsia, and endoscopic findings noted on gross
inspection of the gastric mucosa is poor. Therefore, there is no typical
clinical manifestation of gastritis.
Acute Gastritis
The
most common causes of acute gastritis are infectious. Acute infection with H.
pylori induces gastritis. However, H. pylori acute gastritis has not
been extensively studied. Reported as presenting with sudden onset of
epigastric pain, nausea, and vomiting, limited mucosal histologic studies
demonstrate a marked infiltrate of neutrophils with edema and hyperemia. If not
treated, this picture will evolve into one of chronic gastritis.
Hypochlorhydria lasting for up to 1 year may follow acute H. pylori
infection.
The
highly acidic gastric environment may be one reason why infectious processes of
the stomach are rare. Bacterial infection of the stomach or phlegmonous
gastritis is a rare potentially life-threatening disorder, characterized by
marked and diffuse acute inflammatory infiltrates of the entire gastric wall,
at times accompanied by necrosis. Elderly individuals, alcoholics, and AIDS
patients may be affected. Potential iatrogenic causes include polypectomy and
mucosal injection with India ink. Organisms associated with this entity include
streptococci, staphylococci, Escherichia coli, Proteus, and Haemophilus.
Failure of supportive measures and antibiotics may result in gastrectomy.
Other
types of infectious gastritis may occur in immunocompromised individuals such
as AIDS patients. Examples include herpetic (herpes simplex) or CMV gastritis.
The histologic finding of intranuclear inclusions would be observed in the
latter.
Chronic Gastritis
Chronic
gastritis is identified histologically by an inflammatory cell infiltrate
consisting primarily of lymphocytes and plasma cells, with very scant
neutrophil involvement. Distribution of the inflammation may be patchy,
initially involving superficial and glandular portions of the gastric mucosa.
This picture may progress to more severe glandular destruction, with atrophy
and metaplasia. Chronic gastritis has been classified according to histologic
characteristics. These include superficial atrophic changes and gastric
atrophy.
The
early phase of chronic gastritis is superficial gastritis. The
inflammatory changes are limited to the lamina propria of the surface mucosa,
with edema and cellular infiltrates separating intact gastric glands.
Additional findings may include decreased mucus in the mucous cells and
decreased mitotic figures in the glandular cells. The next stage is atrophic
gastritis. The inflammatory infiltrate extends deeper into the mucosa, with
progressive distortion and destruction of the glands. The final stage of
chronic gastritis is gastric atrophy. Glandular structures are lost;
there is a paucity of inflammatory infiltrates. Endoscopically the mucosa may
be substantially thin, permitting clear visualization of the underlying blood
vessels.
Gastric
glands may undergo morphologic transformation in chronic gastritis. Intestinal
metaplasia denotes the conversion of gastric glands to a small intestinal
phenotype with small-bowel mucosal glands containing goblet cells. The
metaplastic changes may vary in distribution from patchy to fairly extensive
gastric involvement. Intestinal metaplasia is an important predisposing factor
for gastric cancer (Chap. 90).
Chronic
gastritis is also classified according to the predominant site of involvement.
Type A refers to the body-predominant form (autoimmune) and type B is the
central-predominant form (H. pylori-related). This classification is
artificial in view of the difficulty in distinguishing these two entities. The
term AB gastritis has been used to refer to a mixed antral/body picture.
Type A Gastritis
The
less common of the two forms involves primarily the fundus and body, with
antral sparing. Traditionally, this form of gastritis has been associated with
pernicious anemia (Chap. 107) in the presence of circulating antibodies against
parietal cells and intrinsic factor; thus it is also called autoimmune
gastritis. H. pylori infection can lead to a similar distribution of
gastritis. The characteristics of an autoimmune picture are not always present.
Antibodies
to parietal cells have been detected in >90% of patients with pernicious
anemia and in up to 50% of patients with type A gastritis. Anti-parietal cell
antibodies are cytotoxic for gastric mucous cells. The parietal cell antibody
is directed against H+,K+-ATPase. T cells are also
implicated in the injury pattern of this form of gastritis.
Parietal
cell antibodies and atrophic gastritis are observed in family members of
patients with pernicious anemia. These antibodies are observed in up to 20% of
individuals over age 60 and in ~20% of patients with vitiligo and Addison's
disease. About half of patients with pernicious anemia have antibodies to thyroid
antigens, and about 30% of patients with thyroid disease have circulating
anti-parietal cell antibodies. Anti-intrinsic factor antibodies are more
specific than parietal cell antibodies for type A gastritis, being present in
~40% of patients with pernicious anemia. Another parameter consistent with this
form of gastritis being autoimmune in origin is the higher incidence of
specific familial histocompatibility haplotypes such as HLA-B8 and -DR3.
The
parietal cell-containing gastric gland is preferentially targeted in this form
of gastritis, and achlorhydria results. Parietal cells are the source of
intrinsic factor, lack of which will lead to vitamin B12 deficiency
and its sequelae (megaloblastic anemia, neurologic dysfunction).
Gastric
acid plays an important role in feedback inhibition of gastrin release from G
cells. Achlorhydria, coupled with relative sparing of the antral mucosa (site
of G cells), leads to hypergastrinemia. Gastrin levels can be markedly elevated
(>500 pg/mL) in patients with pernicious anemia. ECL cell hyperplasia with
frank development of gastric carcinoid tumors may result from gastrin trophic
effects. The role of gastrin in carcinoid development is confirmed by the
observation that antrectomy leads to regression of these lesions.
Hypergastrinemia and achlorhydria may also be seen in non-pernicious
anemia-associated type A gastritis.
Type B gastritis
Type
B, or antral-predominant, gastritis is the more common form of chronic
gastritis. H. pylori infection is the cause of this entity. Although
described as "antral-predominant," this is likely a misnomer in view
of studies documenting the progression of the inflammatory process towards the
body and fundus of infected individuals. The conversion to a pan-gastritis is
time-dependent-estimated to require 15 to 20 years. This form of gastritis
increases with age, being present in up to 100% of people over age 70.
Histology improves after H. pylori eradication. The number of H.
pylori organisms decreases dramatically with progression to gastric
atrophy, and the degree of inflammation correlates with the level of these
organisms. Early on, with antral-predominant findings, the quantity of H.
pylori is highest and a dense chronic inflammatory infiltrate of the lamina
propria is noted accompanied by epithelial cell infiltration with
polymorphonuclear leukocytes (Fig. 285-12).
Figure 285-12: Chronic gastritis and
Helicobacter pylori organisms. A. H&E stain of gastric mucosa
showing surface foveolar cells, adherent mucus, and scattered bacillary forms
within the mucus. B. Steiner silver stain of superficial gastric mucosa,
showing abundant darkly staining microorganisms layered over the apical portion
of the surface epithelium. Note that there is no tissue invasion.
Multifocal
atrophic gastritis, gastric atrophy with subsequent metaplasia, has been
observed in chronic H. pylori-induced gastritis. This may ultimately
lead to development of gastric adenocarcinoma (Fig. 285-13; Chap. 90). H.
pylori infection is now considered an independent risk factor for gastric
cancer. Worldwide epidemiologic studies have documented a higher incidence of H.
pylori infection in patients with adenocarcinoma of the stomach as compared
to control subjects. Seropositivity for H. pylori is associated with a
three- to sixfold increased risk of gastric cancer. This risk may be as high as
ninefold after adjusting for the inaccuracy of serologic testing in the
elderly. The mechanism by which H. pylori infection leads to cancer is
unknown. However, eradication of H. pylori as a general preventative
measure for gastric cancer is not recommended.
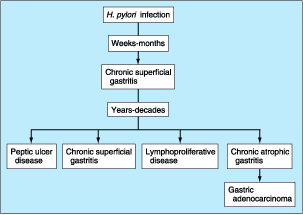
Figure 285-13: Potential long-term
consequences of H. pylori infection.
Infection
with H. pylori is also associated with development of a low grade B cell
lymphoma, gastric MALT lymphoma (Chap. 112). The chronic T cell stimulation
caused by the infection leads to production of cytokines that promote the B
cell tumor. Tumor growth remains dependent upon the presence of H. pylori
in that its eradication is often associated with complete regression of the
tumor. The tumor may take more than a year to regress after treating the
infection. Such patients should be followed by EUS every 2 to 3 months. If the
tumor is stable or decreasing in size, no other therapy is necessary. If the
tumor grows, it may have become a high-grade B cell lymphoma. When the tumor
becomes a high-grade aggressive lymphoma histologically, it loses
responsiveness to H. pylori eradication.
![]() Treatment
Treatment
Treatment
in chronic gastritis is aimed at the sequelae and not the underlying
inflammation. Patients with pernicious anemia will require parenteral vitamin B12
supplementation on a long-term basis. Eradication of H. pylori is not
routinely recommended unless PUD or a low-grade MALT lymphoma is present.
Miscellaneous Forms of Gastritis
Lymphocytic gastritis is characterized histologically by intense
infiltration of the surface epithelium with lymphocytes. The infiltrative
process is primarily in the body of the stomach and consists of mature T cells
and plasmacytes. The etiology of this form of chronic gastritis is unknown. It
has been described in patients with celiac sprue, but whether there is a common
factor associating these two entities is unknown. No specific symptoms suggest
lymphocytic gastritis. A subgroup of patients has thickened folds noted on
endoscopy. These folds are often capped by small nodules that contain a central
depression or erosion; this form of the disease is called varioliform
gastritis. H. pylori probably plays no significant role in
lymphocytic gastritis. Therapy with glucocorticoids or sodium cromoglycate has
obtained unclear results.
Marked
eosinophilic infiltration involving any layer of the stomach (mucosa,
muscularis propria, and serosa) is characteristic of eosinophilic gastritis.
Affected individuals will often have circulating eosinophilia with clinical
manifestation of systemic allergy. Involvement may range from isolated gastric
disease to diffuse eosinophilic gastroenteritis. Antral involvement
predominates, with prominent edematous folds being observed on endoscopy. These
prominent antral folds can lead to outlet obstruction. Patients can present
with epigastric discomfort, nausea, and vomiting. Treatment with
glucocorticoids has been successful.
Several
systemic disorders may be associated with granulomatous gastritis.
Gastric involvement has been observed in Crohn's disease. Involvement may range
from granulomatous infiltrates noted only on gastric biopsies to frank
ulceration and stricture formation. Gastric Crohn's disease usually occurs in
the presence of small-intestinal disease. Several rare infectious processes can
lead to granulomatous gastritis, including histoplasmosis, candidiasis,
syphilis, and tuberculosis. Other unusual causes of this form of gastritis
include sarcoidosis, idiopathic granulomatous gastritis, and eosinophilic
granulomas involving the stomach. Establishing the specific etiologic agent in
this form of gastritis can be difficult, at times requiring repeat endoscopy
with biopsy and cytology. Occasionally, a surgically obtained full-thickness
biopsy of the stomach may be required to exclude malignancy.
Ménétrier's Disease
Ménétrier's
disease is a rare entity characterized by large, tortuous gastric mucosal
folds. The differential diagnosis of large gastric folds includes ZES,
malignancy, infectious etiologies (CMV, histoplasmosis, syphilis), and
infiltrative disorders such as sarcoidosis. The mucosal folds in Ménétrier's
disease are often most prominent in the body and fundus. Histologically,
massive foveolar hyperplasia (hyperplasia of surface and glandular mucous
cells) is noted, which replaces most of the chief and parietal cells. This
hyperplasia produces the prominent folds observed. The pits of the gastric
glands elongate and may become extremely tortuous. Although the lamina propria
may contain a mild chronic inflammatory infiltrate, Ménétrier's disease is not
considered a form of gastritis. The etiology of this unusual clinical picture
is unknown. Overexpression of growth factors such as TGF-![]() may be involved in the process.
may be involved in the process.
Epigastric
pain at times accompanied by nausea, vomiting, anorexia, and weight loss are
signs and symptoms in patients with Ménétrier's disease. Occult
gastrointestinal bleeding may occur, but overt bleeding is unusual and, when
present, is due to superficial mucosal erosions. Between 20 and 100% of
patients (depending on time of presentation) develop a protein-losing
gastropathy accompanied by hypoalbuminemia and edema. Gastric acid secretion is
usually reduced or absent because of the replacement of parietal cells. Large
gastric folds are readily detectable by either radiographic (barium meal) or
endoscopic methods. Endoscopy with deep mucosal biopsy (and cytology) is
required to establish the diagnosis and exclude the other entities that may
present in a similar manner. A nondiagnostic biopsy may lead to a surgically
obtained full-thickness biopsy to exclude malignancy.
![]() Treatment
Treatment
Medical
therapy with anticholinergic agents, prostaglandins, PPIs, prednisone, and H2
receptor antagonists has obtained varying results. Anticholinergics decrease
protein loss. A high-protein diet should be recommended to replace protein loss
in patients with hypoalbuminemia. Ulcers should be treated with a standard
approach. Severe disease with persistent and substantial protein loss may
require total gastrectomy. Subtotal gastrectomy is performed by some; it may be
associated with higher morbidity and mortality secondary to the difficulty in
obtaining a patent and long-lasting anastomosis between normal and hyperplastic
tissues.